Stadium 1–2
Stadium 1 : Nur retinale Teleangiektasien ohne Exsudate.
Stadium 2A: Teleangiektasien mit Exsudaten außerhalb der Fovea.
Stadium 2B: Exsudate reichen bis zur Fovea. Manifeste Sehverschlechterung.

Der Morbus Coats, 1908 von George Coats beschrieben, ist eine idiopathische retinale Gefäßerkrankung. Sie ist gekennzeichnet durch eine abnorme Erweiterung der Netzhautkapillaren (Teleangiektasien) und die Ansammlung von Exsudaten intra- und subretinal aus den Gefäßwänden.
Die Erkrankung ist sporadisch und nicht erblich, ohne Zusammenhang mit systemischen Erkrankungen oder Familienanamnese1). Die Inzidenz beträgt 0,09 pro 100.000 Personen und ist selten2). Etwa 75 % der Patienten sind männlich, 95 % der Fälle sind einseitig, und sie tritt vorwiegend vor dem 20. Lebensjahr (Durchschnittsalter etwa 5 Jahre) auf.
Ein Erwachsenenbeginn ist sehr selten, zeigt aber ein anderes klinisches Bild als die kindliche Form. Erwachsenenfälle sind milder, mit langsamer Progression und gutem Therapieansprechen3). In einem Bericht entwickelten nur 21 % der 48 Augen mit Erwachsenenbeginn eine exsudative Netzhautablösung, verglichen mit 81 % bei kindlichem Beginn3).
Die milde Form wird auch als Leber’sche Milliaraneurysmen bezeichnet und ist Teil des Spektrums des Morbus Coats. Auch die idiopathische makuläre Teleangiektasie Typ 1 wird als Teil desselben Spektrums angesehen.
Es wurden Fälle mit Beginn nach dem 35. Lebensjahr berichtet, die als adulte Form des Morbus Coats anerkannt sind3). Im Vergleich zur kindlichen Form ist die Progression langsamer, die Häufigkeit einer exsudativen Ablösung geringer und das Ansprechen auf die Behandlung oft gut. Der Beginn ist jedoch selten, und bei ähnlichen Befunden bei Erwachsenen ist die Abgrenzung zu anderen Erkrankungen wichtig.
Bei Auftreten im Kleinkindalter äußert das betroffene Kind selten eigene Symptome.
Bei Erwachsenen ist die Sehschärfe relativ gut erhalten, und die Erkrankung wird manchmal zufällig bei einer Vorsorgeuntersuchung entdeckt3).
Charakteristisch für den Fundusbefund sind in der Peripherie konzentrierte, abnorm erweiterte Netzhautgefäße und die Ansammlung gelb-weißer sub- und intraretinaler Exsudate. In der Fluoreszenzangiographie zeigen sich periphere Netzhautgefäßverschlüsse, Teleangiektasien, Mikroaneurysmen und Neovaskularisationen. Im fortgeschrittenen Stadium kommt es zu einer exsudativen Netzhautablösung, die schließlich zu einer totalen Ablösung führen kann. Ultraschall, CT und MRT zeigen eine homogene subretinale Flüssigkeit in allen Bereichen, was ein wichtiger bildgebender Unterschied zum Retinoblastom ist, das ein inhomogenes Bild mit Verkalkungen aufweist.
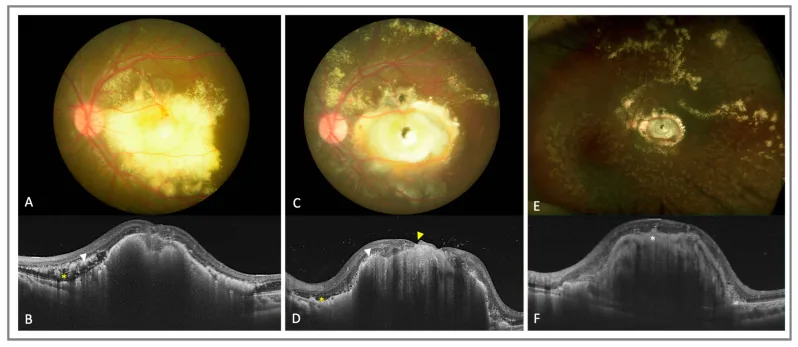
In späteren Stadien treten Vorderabschnittskomplikationen wie Rubeosis iridis und Neovaskularisationsglaukom auf. Ein Makulaloch ist eine seltene Komplikation, die in der Literatur nur in etwa 7 Fällen berichtet wurde4). Der fibröse Knoten ist ein Faktor für eine schlechte Sehprognose, und in der OCTA wurden Typ-3-Neovaskularisationen (SVC→DVC→avaskulärer Komplex) im Knoten bestätigt5).
Die Shields-Klassifikation unterteilt den Morbus Coats in die folgenden 5 Stadien. Sie dient der Wahl der Behandlungsstrategie und der Prognoseabschätzung.
Stadium 1–2
Stadium 1 : Nur retinale Teleangiektasien ohne Exsudate.
Stadium 2A: Teleangiektasien mit Exsudaten außerhalb der Fovea.
Stadium 2B: Exsudate reichen bis zur Fovea. Manifeste Sehverschlechterung.
Stadium 3
Stadium 3A1: Subtotale Netzhautablösung außerhalb der Fovea.
Stadium 3A2: Subtotale Netzhautablösung mit Beteiligung der Fovea. Schlechte Sehprognose.
Stadium 3B: Totale Netzhautablösung. Notfallmäßiger Eingriff erforderlich.
Stadium 4–5
Stadium 4: Totale Netzhautablösung mit sekundärem Glaukom. Kann schmerzhaft sein.
Stadium 5: Endstadium. Phthisis bulbi (Atrophie). Enukleation wird erwogen.
Mehrere Erkrankungen zeigen eine Leukokorie, Morbus Coats ist nur eine davon. Die wichtigste Differentialdiagnose ist das Retinoblastom (Rb), das aufgrund seiner Lebensbedrohlichkeit eine sofortige Abklärung erfordert. Weitere Differentialdiagnosen sind Frühgeborenenretinopathie, persistierendes hyperplastisches primäres Vitreus (PHPV) und Endophthalmitis. Siehe Abschnitt „Diagnose und Untersuchungsmethoden“ für Details.
Die Ursache des Morbus Coats ist unbekannt, es besteht kein Zusammenhang mit systemischen Erkrankungen oder Familienanamnese. Genetische Hintergründe wie Instabilität der Chromosomen 3 und 13 sowie Assoziationen mit den Genen NDP (Morbus Norrie) und CRB1 wurden berichtet, sind aber nicht etabliert.
Als Ausgangspunkt der Pathophysiologie wird die Störung der inneren Blut-Retina-Schranke (iBRB) angenommen 1). Der Verlust von Perizyten (Stützzellen des Gefäßendothels) schwächt die Gefäßwand, was zu abnorm erweiterten Kapillaren und Aneurysmen führt 1). Plasma bestandteile treten aus und sammeln sich in der Gefäßwand und den Netzhautschichten an, was zu einer Wandverdickung und einem Teufelskreis weiterer Exsudation führt 2).
Eine hohe VEGF-Umgebung fördert vermutlich die Erweiterung peripherer Kapillaren 2), was die theoretische Grundlage für die Anti-VEGF-Therapie darstellt.
Die Diagnose wird durch eine Kombination mehrerer Untersuchungen gestellt, wobei die sichere Abgrenzung zum Retinoblastom (Rb) die wichtigste Aufgabe ist.
Die Fundusuntersuchung in Mydriasis zeigt ein geschlängeltes, abnormales Gefäßnetz in der Peripherie und gelb-weiße subretinale Exsudate. Wenn die Exsudate die Makula erreichen, nehmen sie ein hartes, weißliches Aussehen an.
Dies ist eine der wichtigsten Untersuchungen für die Diagnose des Morbus Coats. Charakteristische Befunde sind eine deutliche Erweiterung der Kapillaren, Mikroaneurysmen, arteriovenöse Anastomosen und „Glühbirnen“ (birnenförmige Fluoreszenzlecks). Sie ist unerlässlich für die Abgrenzung des abnormalen Gefäßbereichs und die Festlegung der Laserkoagulationsstellen.
Bestätigt das Fehlen einer soliden Raumforderung. Rb zeigt im B-Mode häufig hyperechogene Echos (Verkalkungen) innerhalb der soliden Raumforderung, während Morbus Coats keine solide Raumforderung bildet.
Beurteilt das Vorhandensein von Verkalkungen. Rb geht häufig mit Verkalkungen einher, Morbus Coats dagegen nicht. Dieser Befund ist ein wichtiges Kriterium für die Differentialdiagnose.
Die wichtigsten Unterscheidungsmerkmale zwischen Morbus Coats und Retinoblastom sind im Folgenden aufgeführt.
| Merkmal | Morbus Coats | Retinoblastom |
|---|---|---|
| Häufigkeitsalter | Etwa 5 Jahre | 1–2 Jahre |
| Geschlechterverteilung | Männlich 75 % | Keine |
| Beidseitigkeit | Etwa 5 % | Etwa 40 % |
| Verkalkung | Nicht vorhanden | Vorhanden (häufig) |
| Solider Tumor | Keiner | Vorhanden |
| Ultraschall | Subretinale Flüssigkeitsansammlung | Solider Tumor, innere Verkalkungen, hinterer Schallschatten |
| MRT | Subretinale Flüssigkeit homogen | Inhomogen (Tumorsignal) |
Weitere Differenzialdiagnosen umfassen retinales Hämangiom, Von-Hippel-Lindau-Krankheit, persistierendes hyperplastisches primäres Vitreum (PHPV), familiäre exsudative Vitreoretinopathie (FEVR), Toxokariose, vasoproliferativer Tumor, Eales-Krankheit usw.
Ziel der Behandlung ist es, die abnormen Gefäße zu verschließen und die Produktion von Exsudat zu stoppen. Es wird ein stufenweiser Ansatz je nach Stadium verfolgt.
Dies ist die Erstlinientherapie. Die mittels FA identifizierten abnorm erweiterten Gefäße und Mikroaneurysmen werden direkt koaguliert, und auch die peripheren Nichtperfusionsbereiche werden photokoaguliert. Bei Kindern wird der Eingriff unter Vollnarkose durchgeführt. Oft sind mehrere Sitzungen erforderlich, und nach der Behandlung werden regelmäßige FA-Nachuntersuchungen und zusätzliche Koagulationen wiederholt.
Dies ist die nächste Option für Läsionen der vorderen Peripherie oder für Stellen, die einer Photokoagulation schwer zugänglich sind. Sie kann auch in Kombination mit der Photokoagulation eingesetzt werden.
Laserkoagulation
Indikationen : Abnorme Gefäße und exsudative Läsionen der Stadien 1 bis 3A.
Methode : Direkte Koagulation der Teleangiektasien und Nichtperfusionsbereiche unter FA-Führung. Bei Kindern unter Vollnarkose.
Merkmale : Wiederholbar. Regelmäßige FA-Nachuntersuchungen und zusätzliche Koagulationen nach der Behandlung sind das Standardmanagement.
Kryokoagulation
Indikationen : Schwer photokoagulierbare vordere periphere Läsionen, Unterstützung bei schweren Fällen bis Stadium 3B.
Methode : Transsklerales Aufsetzen einer Kryosonde zur Koagulation und Okklusion der abnormen Gefäße.
Merkmale : Kann auch bei trüben Medien oder in den äußersten peripheren Bereichen durchgeführt werden.
Vitreoretinale Chirurgie
Indikationen : Stadium 3B (totale Netzhautablösung) oder höher, therapierefraktäre Fälle nach Kryokoagulation.
Methode : Externe Drainage oder Vitrektomie zur Drainage der subretinalen Flüssigkeit und interne Wiederanlage der Netzhaut 8). In schweren Fällen kann eine Drainage der subretinalen Flüssigkeit in Kombination mit einer Cerclage (Sklera-Buckel) durchgeführt werden.
Merkmale : Bei Fällen mit Makulaforamen wurde die Flip-Technik der inneren Grenzmembran (ILM) als wirksam berichtet 4).
Die Anwendung von Anti-VEGF-Medikamenten beim Morbus Coats ist noch nicht konsentiert und wird als adjuvante Therapie in Kombination mit der Photokoagulation betrachtet, nicht als Standardbehandlung.
Ein Fall von adultem Morbus Coats wurde mit einer Kombination aus intravitrealer Injektion von Ranibizumab 0,5 mg und Laserkoagulation behandelt, wobei sich der bestkorrigierte Visus von Fingerzählen auf 20/60 verbesserte3).
In einem Bericht über pädiatrische Fälle, die alle 6 Wochen unter EUA (Untersuchung in Vollnarkose) mit einer Kombination aus intravitrealer Injektion von Bevacizumab 1,25 mg, sub-Tenon-Triamcinolon und Laser behandelt wurden, wurde darauf hingewiesen, dass nach der Behandlung eine paradoxe exsudative Retinopathie auftreten kann2).
Es gibt auch Berichte, dass Brolucizumab bei Bevacizumab-resistenten Fällen wirksam war2). Es wird vermutet, dass eine Anti-VEGF-Therapie zur Prävention der Bildung fibröser Knoten beitragen könnte5).
Bei schmerzhaften blinden Augen (Stadium 4–5) und wenn ein Retinoblastom schwer auszuschließen ist, wird eine Enukleation gewählt.
Beim Morbus Coats kann es über Jahre hinweg zu Rezidiven oder erneuten Ablösungen kommen, und es gibt auch Fälle mit bilateralem Auftreten zu unterschiedlichen Zeitpunkten. Auch nach Abschluss der Behandlung ist eine regelmäßige Neubeurteilung mittels Fluoreszenzangiographie erforderlich. Bei neuen Läsionen sollte eine zusätzliche Photokoagulation oder Kryokoagulation durchgeführt werden.
Derzeit gibt es keinen Konsens über die Verwendung von Anti-VEGF-Medikamenten bei Morbus Coats, und sie gelten nicht als Standardbehandlung. Es häufen sich Berichte über ihren Einsatz als adjuvante Therapie zur Laserphotokoagulation, aber die Bewertung von Wirksamkeit und Sicherheit erfordert weitere Forschung.
Der zentrale Mechanismus des Morbus Coats ist die Störung der inneren Blut-Retina-Schranke (inner blood-retinal barrier; iBRB) 1).
Die iBRB besteht aus den Endothelzellen der Netzhautkapillaren und den sie stützenden Perizyten. Beim Morbus Coats ist die Anzahl der Perizyten deutlich reduziert, was zu einer verminderten vaskulären Endothelstützfunktion führt 1). Auch eine Abnahme der Anzahl der Endothelzellen selbst wurde durch Immunfärbung und Elektronenmikroskopie bestätigt 1).
Die Störung der endothelialen BRB führt zum Austritt und zur Ansammlung von Plasmabestandteilen (hauptsächlich Lipoproteine, Cholesterin) in der Gefäßwand, der Netzhaut und dem subretinalen Raum 1). Die angesammelten Lipide lösen eine Infiltration von lipidhaltigen Makrophagen (Schaumzellen) und eine granulomatöse Immunreaktion aus, die die Gewebeschädigung verschlimmert 1).
Eine hohe VEGF-Umgebung fördert die weitere Erweiterung der peripheren Kapillaren und trägt zum Fortschreiten der Läsion bei 2). Die Beobachtung mittels OCTA hat das Vorhandensein von Typ-3-Neovaskularisationen (gebildet in der Reihenfolge SVC → DVC → avaskulärer Komplex) innerhalb der makulären fibrösen Knoten bei fortgeschrittenen Läsionen bestätigt 5), und das Verständnis des Neovaskularisationsprozesses schreitet voran.
Als Mechanismus für die Entstehung eines Makulaforamens wird angenommen, dass eine periphere Laserbestrahlung zu einer Netzhautverkürzung führt, die eine tangentiale Zugkraft erzeugt und eine Perforation der Makula verursacht 4).
Die OCTA hat eine nicht-invasive Beurteilung der mikrovaskulären Struktur innerhalb fibröser Knoten ermöglicht.
Ong et al. (2021) analysierten die Gefäßstruktur innerhalb makulärer Knoten mittels OCTA im Detail und zeigten das Vorhandensein von Typ-3-Neovaskularisationen, die in der Reihenfolge SVC → DVC → avaskulärer Komplex gebildet werden 5). Diese Erkenntnis ist wichtig für die Aufklärung des Mechanismus der Knotenbildung und als Ziel für die Anti-VEGF-Therapie.
Es wurde über ein paradoxes Ansteigen der Exsudation nach Beginn einer Anti-VEGF-Therapie berichtet, was die Aufklärung des Mechanismus und die Etablierung von Behandlungsstrategien erforderlich macht.
Kalavar et al. (2022) berichteten über einen pädiatrischen Fall von Morbus Coats, bei dem es nach Therapiebeginn zu einer vorübergehenden Exsudationsverschlechterung und Makulasternbildung (macular star formation) kam2). Brolucizumab erwies sich bei einem Bevacizumab-resistenten Fall als wirksam und wird als neue Behandlungsoption diskutiert2).
Nawrocka et al. (2023) berichteten über einen Fall einer Vitrektomie mit der inverted internal limiting membrane flap-Technik bei einem mit Morbus Coats assoziierten Makulaloch4). 18 Monate postoperativ wurde der Verschluss des Makulalochs bestätigt, der bestkorrigierte Visus betrug 20/40. Das mit Morbus Coats assoziierte Makulaloch ist eine seltene Komplikation, von der auf PubMed nur etwa 7 Fälle berichtet wurden4).
Shields et al. (2019) analysierten 351 Augen mit Morbus Coats über 45 Jahre und zeigten eine Verbesserung der Behandlungsergebnisse im Laufe der Jahrzehnte9). Dalvin et al. (2019) analysierten dieselbe Kohorte nach Alterskategorien und zeigten, dass Fälle mit kindlichem Beginn tendenziell schwerwiegender sind und eine schlechtere Sehprognose haben als Fälle mit Erwachsenenbeginn10).
Morbus Coats mit Erwachsenenbeginn ist ein traditionell unterschätztes Krankheitskonzept, aber die Zahl der Berichte nimmt zu3). Die Klärung der klinischen Unterschiede zur kindlichen Form (milder, langsam fortschreitend, gute Therapieansprechen) schreitet voran, und die Etablierung geeigneter Behandlungsprotokolle bei Erwachsenen bleibt eine Herausforderung.