Die Frühgeborenenretinopathie (Retinopathy of Prematurity; ROP) ist eine proliferative Erkrankung der sich entwickelnden Netzhautgefäße.
Die Netzhautgefäße beginnen etwa in der 14. Schwangerschaftswoche von der Papille aus zu entstehen und wachsen nach vorne in den Augenhintergrund. Die oberflächlichen Gefäße erreichen die Peripherie in der 30. Woche, die tiefen Gefäße in der 38.–40. Woche und schließen ihr Wachstum ab. Es gibt individuelle Unterschiede; die temporale Seite ist weiter von der Papille entfernt als die nasale, erreicht die Peripherie später und ist anfälliger für ROP.
Bei Frühgeborenen verbleiben in der peripheren Netzhaut avaskuläre Bereiche. Wenn sich die Umgebung plötzlich von der stabilen Gebärmutter ändert, stoppen die sich entwickelnden Gefäße ihr Wachstum an der Spitze mit den unreifsten Zellen und proliferieren in abnormale Richtungen. Der Mechanismus ist die Freisetzung von VEGF (vaskulärer endothelialer Wachstumsfaktor) aus den avaskulären Bereichen.
Die Phase der fortschreitenden Proliferation wird als aktive Phase bezeichnet; nach der Beruhigung bilden sich die Gefäßkomponenten zurück und hinterlassen Narben wie fibröses Bindegewebe, Netzhauttraktion oder -degeneration, was als Narbenstadium bezeichnet wird.
Die Häufigkeit und Schwere der ROP ist umso höher, je unreifer das Wachstum der Netzhautgefäße ist. Je niedriger das Gestationsalter und das Geburtsgewicht, desto höher die Inzidenz und desto schwerwiegender der Verlauf. Hochkonzentrierter Sauerstoff ist der größte auslösende Faktor, der die ROP verschlimmert; weitere Faktoren sind Atemnotsyndrom, Austauschtransfusion, Sepsis, intraventrikuläre Blutung, chirurgische Eingriffe und ein Ungleichgewicht von Ernährung oder Flüssigkeitszufuhr.
Erstmals 1942 von Terry beschrieben, führte Nagata 1967 die weltweit erste Photokoagulationsbehandlung durch, die sich zur Standardtherapie entwickelte. Historisch gab es drei Epidemien der Frühgeborenenretinopathie 1): die erste (1940er-1950er Jahre) verursacht durch hochkonzentrierte Sauerstoffgabe im Inkubator, die zweite (1970er-1980er Jahre) aufgrund der verbesserten Überlebensrate extrem untergewichtiger Frühgeborener und die dritte, derzeit andauernde, in Ländern mit niedrigem und mittlerem Einkommen mit begrenzten medizinischen Ressourcen 1). Der Anteil der ROP an den Ursachen für Erblindung im Kindesalter betrug 1990 etwa 10 %, ist aber heute auf 30 % gestiegen. Weltweit wird geschätzt, dass jährlich etwa 184.700 Frühgeborene eine ROP entwickeln und etwa 20.000 Kinder erblinden oder eine schwere Sehbehinderung erleiden 1). Bei extrem untergewichtigen Frühgeborenen (< 1.000 g) entwickeln 86,1 % eine ROP, und 41 % der Fälle benötigen eine Behandlung. Die Inzidenzrate der ROP in den USA stieg von 4,4 % im Jahr 2004 auf 8,1 % im Jahr 2019 1).
Die ungefähren Inzidenzraten sind wie folgt:
Zielgruppe
Inzidenzrate
Extrem untergewichtige Frühgeborene (< 1.000 g), Japan
86,1 %
Gestationsalter ≤ 27 Wochen, USA
89,0 %
Gestationsalter 27–31 Wochen, USA
51,7 %
Gestationsalter ≥ 32 Wochen, USA
14,2 %
Alle Geburten (gesamt), USA
0,12 %
Die wichtigsten Risikofaktoren sind unten aufgeführt 1).
Risikofaktoren
Beschreibung
Gestationsalter < 32 Wochen
Einer der wichtigsten Risikofaktoren
Geburtsgewicht < 1,5 kg
Einer der wichtigsten Risikofaktoren
Hochkonzentrierte und langfristige Sauerstoffgabe
Hauptauslöser der Erkrankung
Mehrlingsschwangerschaft
Führt zu niedrigem Geburtsgewicht
Atemnotsyndrom (NRDS)
Schwere Fälle, die eine Beatmung erfordern
Sepsis / intraventrikuläre Blutung
Systemische Entzündung / Kreislaufstörungen
Verzögerte postnatale Gewichtszunahme
Assoziiert mit niedrigem IGF-1-Spiegel
Transfusion / Erythropoetin-Gabe
Veränderung der Sauerstofftransportkapazität
QBei welchen Babys tritt die Frühgeborenenretinopathie am häufigsten auf?
A
Je kürzer die Gestationswoche und je niedriger das Geburtsgewicht, desto höher das Risiko, an der Erkrankung zu erkranken. Insbesondere Frühgeborene mit einem Gestationsalter unter 32 Wochen und einem Geburtsgewicht unter 1.500 g bilden die Hauptrisikogruppe. Auch postnatale Umweltfaktoren wie die Gabe von hochkonzentriertem Sauerstoff tragen zur Entstehung bei. Das Zusammentreffen mehrerer Risikofaktoren erhöht die Wahrscheinlichkeit eines schweren Verlaufs.
Da die akute Phase der Frühgeborenenretinopathie in der Neugeborenen- und Säuglingszeit auftritt, kann das betroffene Kind selbst keine Symptome äußern. Eltern oder medizinisches Personal können die folgenden Anzeichen bemerken.
Leukokorie (weiße Pupille) : In fortgeschrittenen Fällen (Stadium 4–5) führt eine Netzhautablösung dazu, dass die Pupille weiß erscheint.
Strabismus (Schielen) : Zeigt eine schlechte Sehschärfe oder ein Ungleichgewicht der binokularen Sehfunktion an.
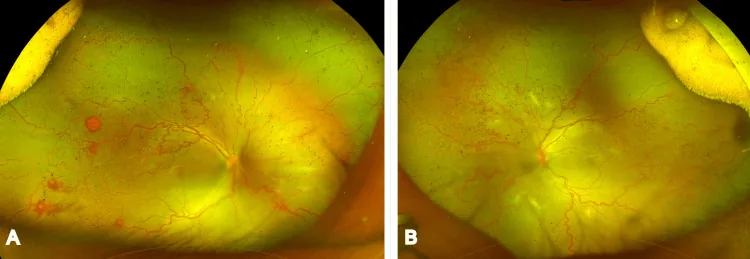
Jain KS, et al. Zone 3 Aggressive Retinopathy of Prematurity in a Near-Term Delivered Big Baby With a Birth Weight of 3,200 g. Cureus. 2026. Figure 1. PMCID: PMC13016036. License: CC BY.
Ultraweitwinkel-Fundusfotografien beider Augen zeigen eine deutliche Erweiterung und Schlängelung der Netzhautgefäße, abnorme Gefäße und Blutungen in der peripheren Netzhaut. Dies sind klinische Befunde einer hohen Aktivität der Frühgeborenenretinopathie. Sie entsprechen der Stadieneinteilung, die im Abschnitt „Hauptsymptome und klinische Befunde“ beschrieben wird.
In Japan wurde 1975 die Klassifikation des Gesundheitsministeriums erstellt und 1983 überarbeitet. Sie unterteilt die ROP in Typ I (klassische ROP), die schrittweise fortschreitet, und Typ II (fulminant), die schnell zu einer Netzhautablösung führt. Die internationale Klassifikation (ICROP) hat bei ihrer Überarbeitung im Jahr 2005 die Konzepte der Klassifikation des Gesundheitsministeriums vollständig übernommen, sodass der Inhalt nahezu identisch ist. Die neueste ICROP, die dritte Auflage (ICROP3), wurde 2021 veröffentlicht2).
Entsprechung zwischen der Klassifikation des Gesundheitsministeriums und ICROP3
Klassifikation des Gesundheitsministeriums
Internationale Klassifikation (ICROP3)
Typ I Stadium 1: intraretinale Neovaskularisation
Stadium 1: Demarkationslinie
Typ I Stadium 2: Bildung einer Demarkationslinie
Stadium 2: Leiste (ridge)
Typ I Stadium 3: intravitreale Exsudation und Proliferation (früh/mittel/spät)
Die Lokalisation der Läsionen wird durch die Zonen I bis III ausgedrückt. Zone I ist der Bereich innerhalb eines Kreises, der um die Papille zentriert ist und dessen Radius das Doppelte des Abstands zwischen Papille und Fovea beträgt. Dies entspricht dem sichtbaren Bereich, wenn der Rand einer +25 D oder +28 D Beobachtungslinse am gegenüberliegenden Rand der Papille platziert wird. Zone II ist der Bereich außerhalb von Zone I, innerhalb eines Kreises mit einem Radius von der Papille bis zur nasalen Ora serrata. Zone III ist der temporale Mondsichelbereich außerhalb von Zone II. Je weiter posterior die Läsion (Zone I), desto höher der Schweregrad. In der ICROP3 wurde das Konzept der posterioren Zone II (ein 2-Papillendurchmesser breiter Streifen von der Zone-I-Grenze) hinzugefügt 2).
Stadium 1 (Demarkationslinie) : Am vorderen Ende des Gefäßwachstums bildet sich eine weiße Demarkationslinie in der Netzhaut.
Stadium 2 (Leiste) : Die Proiferation der teppichartigen mesenchymalen Zellen im vorderen Bereich wird dicker, und die Demarkationslinie wölbt sich in den Glaskörperraum vor und bildet eine Leiste.
Stadium 3 (extraretinale fibrovaskuläre Proliferation) : Die Gefäßbüschel verschmelzen bogenförmig, intravitreale Neovaskularisationen bilden Gefäßlumen, und das umgebende Bindegewebe produziert Kollagen. Je nach Ausprägung der Befunde wird in mild, moderat und schwer unterteilt. In der ICROP3 wurde das Konzept der flachen extraretinalen Neovaskularisation ohne Leiste in Stadium 3 aufgenommen 2).
Stadium 4 (partielle Netzhautablösung) : Das Bindegewebe innerhalb der fibrovaskulären Proliferation kontrahiert und zieht an der Netzhaut, was zu einer partiellen Netzhautablösung führt. Es wird in 4A (Makula nicht betroffen) und 4B (Makula betroffen) eingeteilt.
Stadium 5 (totale Netzhautablösung) : Die fibrovaskuläre Proliferation ist ausgedehnt und übt starken Zug aus, was zu einer totalen Netzhautablösung führt. In der ICROP3 wurde Stadium 5 in die folgenden drei Subtypen eingeteilt 2).
Subtyp
Merkmale
5A
Die Papille ist im Ophthalmoskop sichtbar (offene Trichterablösung)
5B
Retrolentikuläres fibrovaskuläres Gewebe oder geschlossener Trichter, der die Papille unsichtbar macht
In den Stadien 1 bis 3 wird eine Plus-Erkrankung definiert, wenn eine Dilatation der Netzhautvenen und eine Tortuosität der Arterien in zwei oder mehr hinteren Quadranten vorliegt. In der ICROP3 wird die Plus-Erkrankung als ein kontinuierliches Spektrum von normal über Pre-Plus bis hin zu Plus betrachtet 2). Die Beurteilung der Plus-Erkrankung erfolgt an den Gefäßen in Zone I.
In der ICROP3 wurde die frühere AP-ROP (aggressive posteriore ROP) in A-ROP umbenannt. Diese Definition wurde erweitert, um Fälle außerhalb der posterioren Region, bei größeren Frühgeborenen oder in Regionen mit begrenzten Ressourcen einzuschließen 2).
Die Merkmale der A-ROP sind wie folgt: Sie tritt in der posterioren Region (häufig Zone I, manchmal auch posteriore Zone II) auf, mit ausgeprägter Plus-Erkrankung, zirkumferentieller Beteiligung und Shuntbildung. Die Grenze zwischen vaskularisierter und avaskulärer Netzhaut ist unscharf, und die intravitrealen Neovaskularisationen sind flach, semitransparent und schwer zu erkennen. Sie zeigt nicht die übliche schrittweise Progression von Stadium 1 zu 3 und erreicht unbehandelt schnell Stadium 5. Es besteht ein ausgedehnter Mangel an Kapillarnetzwerk einschließlich des hinteren Pols, und eine weitreichende VEGF-Freisetzung ist die Grundlage der raschen Progression.
Wenn in frühen Läsionen Shunts an den Spitzen der Netzhautgefäße oder Netzhautblutungen zu sehen sind, sollte sofort eine Photokoagulation durchgeführt werden.
Die Entstehung der Frühgeborenenretinopathie beruht auf einer Wechselwirkung zwischen dem unreifen retinalen Gefäßsystem und der Sauerstoffumgebung sowie dem Allgemeinzustand nach der Geburt. Hochkonzentrierter Sauerstoff ist der größte auslösende Faktor, der die ROP verschlimmert. Darüber hinaus sind das Atemnotsyndrom, Austauschtransfusionen, Sepsis, intraventrikuläre Blutungen, chirurgische Eingriffe in der Vorgeschichte sowie ein Ungleichgewicht von Ernährung und Flüssigkeitszufuhr auf komplexe Weise beteiligt.
Risikofaktor
Beschreibung
Gestationsalter
<32 Wochen: hohes Risiko
Geburtsgewicht
<1.500 g: hohes Risiko
Sauerstoffgabe
Hohe Konzentration und langfristige Gabe
Niedriger IGF-1-Spiegel
Postnatale Mangelernährung und Erkrankungen 1)
Sepsis, Anämie, Transfusion
Verschlechterung des Allgemeinzustands
Atemnotsyndrom
Schwer krankes Kind, das eine Beatmung benötigt
Mehrlingsgeburt
Der kleinere Zwilling hat häufiger eine fortgeschrittene ROP1)
Schlechte Gewichtszunahme nach der Geburt
Grundlage der WINROP/G-ROP-Algorithmen1)
IGF-1 (Insulinähnlicher Wachstumsfaktor-1) ist ein essentieller Signalstoff für die Entwicklung der Netzhautgefäße. Ein niedriger IGF-1-Spiegel nach einer Frühgeburt fördert den Stillstand des Gefäßwachstums1).
Die Entwicklung der Netzhautgefäße beginnt etwa in der 14. Schwangerschaftswoche und ist vor der Geburt abgeschlossen, wenn sie die äußerste Peripherie erreicht haben. Die oberflächlichen Gefäße erreichen die Peripherie in der 30. Woche, die tiefen Gefäße in der 38.–40. Woche. Bei Frühgeborenen verbleiben bei der Geburt avaskuläre Bereiche in der peripheren Netzhaut.
Die Pathologie der ROP wird in zwei Phasen beschrieben1).
Phase 1 (Stillstand der Gefäßentwicklung): Wenn die unreife Netzhaut eines Frühgeborenen einer hyperoxischen Umgebung (außerhalb der Gebärmutter) ausgesetzt wird, werden VEGF und IGF-1 über Sauerstoffsensoren unterdrückt. Dadurch stoppt die normale Entwicklung der Netzhautgefäße und es bildet sich eine avaskuläre Zone.
Phase 2 (Gefäßproliferationsphase): Mit der metabolischen Reifung der avaskulären Netzhaut wird VEGF übermäßig produziert, um den ischämischen Zustand auszugleichen. Dieser VEGF-Überschuss induziert eine abnormale Proliferation des Gefäßendothels, was zur Bildung von fibrovaskulärem Proliferationsgewebe in den Glaskörperraum führt (Stadium 3 und darüber).
Neovaskularisation wächst entlang der Fasern des Glaskörpers in der Glaskörperhöhle und produziert umliegendes Bindegewebe wie Kollagen. Wenn sich dieses Bindegewebe zusammenzieht und die anhaftende Netzhaut zieht, kommt es zu einer Netzhautablösung, die zu schwerer Sehbehinderung oder Erblindung führt.
Bei A-ROP besteht ein ausgedehnter Mangel des Kapillarnetzes einschließlich des hinteren Pols, und aufgrund der Freisetzung von VEGF aus einem großen Bereich einschließlich des hinteren Pols schreitet die Erkrankung schnell voran.
IGF-1 (Insulinähnlicher Wachstumsfaktor-1) ist wichtig für die Koordination der vaskulären und neuronalen Entwicklung der Netzhaut, und ein niedriger IGF-1-Spiegel verzögert die normale Gefäßentwicklung und erhöht die Anfälligkeit für ROP1). Eine verzögerte postnatale Gewichtszunahme ist ebenfalls mit niedrigem IGF-1 verbunden und stellt ein Risiko dar1).
Crunch-Syndrom: Wenn VEGF nach einer Anti-VEGF-Therapie unterdrückt wird, führt der relative Anstieg von TGF-β (fibrosefördernder Faktor) zu einem Ungleichgewicht des VEGF-TGF-β-Gleichgewichts, was zu einer schnellen Kontraktion der fibrovaskulären Membran führt. Dadurch besteht das Risiko einer Verschlechterung der traktiven Netzhautablösung1).
Die Screening-Kriterien auf der Neugeborenen-Intensivstation (NICU) sind Säuglinge mit einem Gestationsalter von weniger als 34 Wochen oder einem Geburtsgewicht von 1.800 g oder weniger. Darüber hinaus gelten Säuglinge mit Sauerstofftherapie, mechanischer Beatmung, Bluttransfusion, Sepsis, intraventrikulärer Blutung, schweren Atem- oder Kreislaufstörungen, Operation unter Vollnarkose oder Hydrops fetalis als Hochrisikogruppe und benötigen auch dann eine Fundusuntersuchung, wenn sie diese Kriterien nicht erfüllen.
Der Zeitpunkt der ersten Untersuchung ist wie folgt:
Die Behandlungsindikationen basieren auf den Kriterien für Typ-1-ROP der ETROP-Studie 1). Wenn eines der folgenden Kriterien erfüllt ist, wird eine Behandlung innerhalb von 72 Stunden nach der Diagnose empfohlen.
① Jede ROP in Zone I mit Plus-Erkrankung
② ROP in Zone I Stadium 3 ohne Plus-Erkrankung
③ ROP in Zone II Stadium 3 mit Plus-Erkrankung
④ A-ROP (so bald wie möglich durchführen)
Alle anderen Fälle werden als Typ-2-ROP eingestuft und beobachtet. ROP in Zone II Stadium 2 mit Plus-Erkrankung wird je nach verwendetem Medikament unterschiedlich behandelt (in der RAINBOW-Studie ausgeschlossen, aber in der FIREFLEYE-Studie eingeschlossen) 3).
Die empfohlenen Untersuchungsintervalle je nach Befund sind unten aufgeführt 1).
Befund
Nächste Untersuchung
Unreife Gefäße in Zone I oder Stadium 1-2, unreife Gefäße in posteriorer Zone II, Verdacht auf A-ROP
1 Mal pro Woche
Unreife Netzhaut in posteriorer Zone II, Zone II Stadium 2, Regression in Zone I
1–2 Wochen
Zone I Stadium 1, unreife Gefäße in Zone II (keine ROP), Regression in Zone II
2 Wochen
Zone III Stadium 1-2, Zone III in Regression
2–3 Wochen
Die Kriterien für das Ende des Screenings sind eine vollständige Vaskularisation bis Zone III oder das Fehlen einer Typ-1-ROP bei einem korrigierten Gestationsalter von 45 Wochen. Nach einer Anti-VEGF-Therapie ist eine Nachbeobachtung bis mindestens zur 65. Woche des korrigierten Gestationsalters erforderlich 1).
Midrin® P oder eine Mischung aus Neosynephrin®, Midrin® P und Cyclogyl® im Verhältnis 2:1:1 (modifizierte Capto-Methode) wird ab einer Stunde vor der Untersuchung dreimal im Abstand von 10 Minuten getropft, um die Pupille zu erweitern.
Weitwinkel-Funduskamera (RetCam usw.) : Ermöglicht eine großflächige Fundusfotografie unter Mydriasis. Wird auch für die Telemedizin verwendet.
KI-gestützte Bilddiagnostik : Die automatische Erkennung der Plus-Erkrankung hat eine Genauigkeit gezeigt, die der von Augenärzten entspricht oder diese übertrifft 1). Ein auf Deep-Learning-Modellen basierender VSS (Vascular Severity Score) wurde als objektiver Indikator für die Diagnose der Plus-Erkrankung entwickelt 1).
Vorhersagemodelle (G-ROP, WINROP) : Erreichen eine Sensitivität von 100 % für die Vorhersage einer Typ-1-ROP basierend auf Gestationsalter, Geburtsgewicht und postnataler Gewichtszunahme 1).
FIRST-ROP-Algorithmus : Schlägt vor, den Beginn des Screenings bei Kindern mit mittlerem Risiko (Gestationsalter ≥ 27 Wochen und Geburtsgewicht ≥ 800 g) auf die 34. Woche des korrigierten Gestationsalters zu verschieben 1).
Fluoreszenzangiographie : Nützlich zur Beurteilung des Vorhandenseins und des Ausmaßes eines Wiederauftretens. Ein Einsatz für die PAR-Bewertung wird erwartet 3).
QWann und wie oft werden die Screening-Untersuchungen durchgeführt?
A
Bei einem Gestationsalter unter 26 Wochen beginnt die erste Untersuchung in der 29. Woche des korrigierten Gestationsalters, bei 26 Wochen oder mehr beginnt sie 3 Wochen nach der Geburt. Danach wird sie je nach Befund alle 1–3 Wochen wiederholt. Nach einer Anti-VEGF-Behandlung wird eine Nachbeobachtung bis zur 65. Woche des korrigierten Gestationsalters empfohlen 3). Das Screening endet, wenn die Netzhautvaskularisation ohne behandlungsbedürftige Befunde abgeschlossen ist.
Das hängt vom Gestationsalter bei der Geburt ab. Bei einem Gestationsalter unter 26 Wochen beginnt die erste Fundusuntersuchung ab der 29. korrigierten Schwangerschaftswoche, bei 26 Wochen oder mehr ab der 3. Lebenswoche. Die Screening-Kriterien sind ein Gestationsalter unter 34 Wochen oder ein Geburtsgewicht von 1.800 g oder weniger, aber bei Risikofaktoren wie Sauerstofftherapie, Transfusion oder Sepsis ist eine Untersuchung auch außerhalb dieser Kriterien erforderlich.
Seit Nagata 1967 die weltweit erste Photokoagulationsbehandlung durchführte, hat sich diese als Standardtherapie für ROP etabliert. Die Photokoagulation des gesamten avaskulären Bereichs ist die Grundlage und wird unter indirekter Ophthalmoskopie durchgeführt. Bei ausgeprägter Linsenvaskularmembran oder Pupillenstarre kann die Durchführung schwierig sein. Die Koagulation ist zeitaufwändig und erfordert Erfahrung des Operateurs; ausgedehnte Koagulation kann zu Gesichtsfeldeinschränkung und Myopie führen.
In der CRYO-ROP-Studie betrugen die schlechten strukturellen Ergebnisse nach einem Jahr 25,7 % in der Kryotherapiegruppe gegenüber 47,4 % in der Beobachtungsgruppe, und der Unterschied blieb nach 15 Jahren signifikant (30 % vs. 52 %)8). In der ETROP-Studie reduzierte die Frühbehandlung die schlechten strukturellen Ergebnisse von 9,1 % auf 15,6 %9).
Da VEGF an der retinalen Neovaskularisation beteiligt ist, wird die intravitreale Injektion von Anti-VEGF-Medikamenten bei ROP versucht. Stand Dezember 2022 sind in Japan die folgenden beiden Anti-VEGF-Medikamente zur ROP-Behandlung zugelassen3).
Ranibizumab (Lucentis®): 0,2 mg/Dosis (0,02 ml). Im November 2019 in Japan zugelassen.
Aflibercept (Eylea®): 0,4 mg/Dosis (0,01 ml). Im September 2022 in Japan zugelassen.
Beide sind nur als Durchstechflaschenpräparate für ROP zugelassen; das Wiederholungsintervall beträgt laut Packungsbeilage mindestens einen Monat3). Bevacizumab ist in Japan und international nicht für ROP zugelassen3).
Ergebnisse der wichtigsten klinischen Studien
Studienname
Population / Medikament
Wichtigste Ergebnisse
BEAT-ROP
Zone I Stadium 3+, Bevacizumab 0,625 mg
Rezidiv in Zone I: 6 % vs. Laser 42 %5, 1)
RAINBOW
Geburtsgewicht < 1.500 g, Ranibizumab 0,2 mg
Behandlungserfolgsrate 80,0 % vs. Laser 66,2 %. Hochgradige Myopie im Alter von 2 Jahren: 5 % vs. 20 %6, 1)
Ranibizumab zeigt 14 Tage nach Injektion keine nachweisbare Senkung des VEGF im Blut, was auf eine geringe systemische Wirkung hindeutet. Bei Aflibercept fällt das freie Aflibercept im Plasma etwa 8 Wochen nach Injektion auf die untere Bestimmungsgrenze ab3).
Kinderspezifische Injektionstechnik3):
Einstich 1,0–1,5 mm hinter dem Limbus (Achtung: abweichend von 3–4 mm bei Erwachsenen)
Da die Linse relativ groß ist, nach unten (hinten) einstechen. Ein Einstich in Richtung Augenmitte birgt das Risiko einer Linsenverletzung
Verwendung einer 30-Gauge-Nadel oder dünner
Durchführung im Inkubator der Neugeborenen-Intensivstation oder im Operationssaal
Anästhesie: Tropfanästhesie, intravenöse oder Vollnarkose je nach Einrichtung wählen
Die Anti-VEGF-Therapie hat drei Hauptziele: Erstens die adjuvante Therapie (Zeitgewinn vor Vitrektomie), zweitens die Salvage-Therapie (Verhinderung einer Netzhautablösung bei Versagen der Photokoagulation), drittens die Monotherapie (Einzelgabe als Alternative zur Photokoagulation). Nach Monotherapie beruhigt sich die ROP und es wachsen Gefäße in die avaskulären Bereiche, aber ein langfristiges Wiederauftreten der Proliferation ist möglich (schwelende Retinopathie).
Der Beobachtungsplan nach einer Anti-VEGF-Therapie ist wie folgt 3).
Erstes Jahr nach der Injektion: möglichst alle 2 Wochen eine Fundusuntersuchung
Nach zusätzlicher Laserbehandlung oder Gefäßentwicklung bis Zone III: alle 2–3 Monate
Bei A-ROP: 2-mal pro Woche bis 2–3 Wochen nach der Injektion, 1-mal pro Woche bis etwa 4 Monate, danach alle 1–2 Wochen
Die Entscheidung über ein Wiederaufflammen basiert auf dem erneuten Auftreten von Plus Disease, und eine zusätzliche Behandlung erfolgt gemäß den ETROP-Kriterien.
Die Fluoreszenzangiographie ist nützlich, um das Vorhandensein und Ausmaß einer erneuten Gefäßproliferation zu beurteilen.
Bei Netzhautablösung ab Stadium 4 ist eine chirurgische Behandlung erforderlich. Bei Typ I/klassischer ROP wird eine linsenschonende Vitrektomie (LSV) durchgeführt, die eine gute Wiederanlegungsrate und Sehprognose bietet. Eine frühe Operation im Stadium 4A beeinflusst die Sehprognose. Bei Typ II/A-ROP ist die LSV aufgrund der hohen Aktivität der fibrovaskulären Proliferation weniger wirksam, und eine Linsenentfernung ist oft erforderlich.
Die anatomische Erfolgsrate der LSV wird für Stadium 4A mit 74–91 %, für Stadium 4B mit 62–92 % und für Stadium 5 mit 22–48 % angegeben 1). Die erwartete Sehschärfe beträgt nach Wiederanlegung im Stadium 4A 20/80 oder besser, nach Reparatur im Stadium 4B Gehsehvermögen und nach Reparatur im Stadium 5 Handbewegungen 1). Die Kataraktbildung nach LSV ist innerhalb von 10 Jahren selten, tritt aber, wenn sie auftritt, meist im ersten postoperativen Jahr auf 1).
QSollte eine Anti-VEGF-Therapie oder eine Laserphotokoagulation gewählt werden?
A
Die Wahl hängt von der Lokalisation und Schwere der Läsion ab. Gemäß dem Leitfaden zur Anti-VEGF-Therapie (2. Auflage) ist die Anti-VEGF-Therapie für Zone I und A-ROP vorteilhaft, während Laser für Zone-II-Läsionen weiterhin eine wichtige Option darstellt 3). Die Anti-VEGF-Therapie hat bei schweren Fällen in Zone I Vorteile wie einfache Verabreichung, kurze Behandlungszeit und geringere Belastung für das Kind, birgt jedoch ein höheres Risiko für ein Wiederaufflammen und erfordert eine langfristige regelmäßige Nachsorge. Die Wahl wird getroffen, nachdem der Familie die Vor- und Nachteile jeder Option erläutert wurden.
QWie häufig treten Wiederaufflammen nach einer Anti-VEGF-Therapie auf?
A
Dies variiert je nach Medikament. Für Aflibercept wurden Wiederaufflammen in 13,9–28 % der Fälle berichtet, für Ranibizumab in 20,8–83,0 %; in der RAINBOW-Studie benötigten 31 % eine zusätzliche Behandlung 3, 1). Bei A-ROP benötigen 75,0–87,5 % eine zusätzliche Behandlung, und eine strenge frühe Überwachung nach der Injektion ist unerlässlich.
In ICROP3 wurden die Konzepte der Regression und Reaktivierung offiziell definiert 2). Die Regression wird in vollständige und unvollständige Regression unterteilt. Nach Anti-VEGF beginnt die Regression der Gefäßveränderungen innerhalb von 1–3 Tagen, nach Laser hingegen dauert es 7–14 Tage. Frühe Anzeichen der Regression sind die Besserung der Plus-Erkrankung und die Gefäßausdehnung in die peripheren avaskulären Bereiche.
Nach unvollständiger Regression kann eine persistierende avaskuläre Retina (PAR) zurückbleiben. PAR tritt nach Anti-VEGF häufiger und ausgedehnter auf als nach Laser oder spontaner Regression 2). Eine Reaktivierung tritt häufiger nach Anti-VEGF-Therapie auf, mit einem Höhepunkt in der korrigierten 37. bis 60. Woche. Sie kann je nach Medikament und Dosis verzögert auftreten 2).
Risikofaktoren für eine Netzhautablösung waren ein Gestationsalter ≤ 29 Wochen (P < 0,05) und eine Vaskularisation bis zur posterioren Zone 2 (P = 0,009) 4). 86,4 % der Netzhautablösungen waren rhegmatogen oder gemischt, und 57,9 % traten vor dem 30. Lebensjahr auf 4). 20 % der abgelösten Augen (28/140) wurden beim ersten Besuch als irreparabel eingestuft 4). Eine unvollständige Vaskularisation (Zone 3 nicht erreicht) wurde bei 71,6 % festgestellt 4).
Diese Ergebnisse legen eine regelmäßige Untersuchung und Beurteilung mittels Ultraweitwinkel-Fluoreszenzangiographie bei unbehandelter ROP nahe 4).
Hohe Myopie nach Laserphotokoagulation tritt bei 20 % der Kinder im Alter von 2 Jahren und bei 24 % nach 5 Jahren auf. In der Ranibizumab-0,2-mg-Gruppe war sie mit 5 % im Alter von 2 Jahren und 8 % nach 5 Jahren signifikant seltener 1). Frühgeborene haben unabhängig von der ROP ein Risiko für hohe Myopie und Makulaanomalien (Verkleinerung der fovealen avaskulären Zone, Abflachung oder Verschwinden der fovealen Vertiefung).
Bei unbehandelter spontaner Regression tritt ein erhöhter Augeninnendruck in 23,2 % der Fälle auf, bei alleiniger Koagulationstherapie in 23,3 % und nach Akutoperation in 58,5 % 1). Im Stadium 5 zeigen 66,7 % und bei aphaken Augen 69,8 % einen erhöhten Augeninnendruck1). 10,0 % der fortgeschrittenen ROP (Stadium 4-5) entwickeln innerhalb von 3 Jahren ein Glaukom, wobei Stadium 5 ein 6,76-faches Risiko im Vergleich zu Stadium 4A und die Linsenentfernung ein 2,76-faches Risiko darstellt 1).
QKann eine unbehandelte ROP im Erwachsenenalter Probleme verursachen?
A
Ja, das ist möglich. Eine multizentrische Studie zu unbehandelter ROP zeigte eine hohe Rate an Spätkomplikationen im Erwachsenenalter, darunter Gitterdegeneration (54 %), Netzhautrisse (30,6 %) und Netzhautablösung (38,6 %) 4). 57,9 % der Netzhautablösungen treten vor dem 30. Lebensjahr auf, was eine lebenslange regelmäßige Fundusuntersuchung unerlässlich macht.
QSollte man sich bei Kindern mit ROP Sorgen um zukünftige Kurzsichtigkeit machen?
A
Dies ist eine besorgniserregende Komplikation. In der Lasergruppe trat nach 5 Jahren bei 24 % eine hohe Myopie auf, während dies in der Ranibizumab-0,2-mg-Gruppe mit 8 % signifikant seltener war 1). Zudem ist die Frühgeburt selbst ein unabhängiger Risikofaktor für hohe Myopie und Makulaanomalien. Eine Nachsorge mit Brillenverordnung und regelmäßigen Refraktionsuntersuchungen ist wichtig.
Der optimale Zeitpunkt der Laserphotokoagulation für PAR nach Anti-VEGF-Therapie ist noch nicht etabliert 1). Eine retrospektive Studie deutet darauf hin, dass eine prophylaktische Laserbehandlung 60 Wochen nach der Korrektur nach Bevacizumab mit einer Verringerung schlechter struktureller Ergebnisse verbunden ist 1). Die Bewertung des Reaktivierungsrisikos mittels Fluoreszenzangiographie (60 Wochen nach Korrektur) wurde als nützlich berichtet 1).
Orale Propranolol: Mit 2 mg/kg/Tag wurde ein präventiver Effekt auf die Progression von ROP im Stadium 2 berichtet, jedoch mit dem Risiko kardiorespiratorischer Komplikationen 1). Topisches Propranolol 0,2 % Augentropfen, beginnend im Stadium 1, wurde als sicher und wirksam berichtet 1).
AA/DHA-Supplementierung: Die Mega Donna Mega Trial (206 Patienten, Gestationsalter < 28 Wochen) berichtete eine Inzidenz schwerer ROP von 15,8 % in der AA+DHA-Gruppe gegenüber 33,3 % in der Kontrollgruppe (50 % Reduktion) 1).
Koffein: Koffein, das bei Frühgeborenenapnoe verabreicht wird, könnte das Fortschreiten der ROP durch Herunterregulierung von VEGF und MMP hemmen 1).
Conbercept (KH902): In China zugelassen. Es zeigte eine ähnliche Wirksamkeit wie Ranibizumab mit einer Rezidivrate von 16,7 % gegenüber 23,3 % (Ranibizumab) 1).
Deep-Learning-Modelle zur automatischen Erkennung der Plus-Krankheit erreichen eine Genauigkeit, die der von Spezialisten entspricht 1). Der vaskuläre Schweregrad-Score (VSS) wurde als objektiver quantitativer Indikator für den Schweregrad der Plus-Krankheit entwickelt 1). Telemedizinprogramme wie SUNDROP ermöglichen die Ausweitung des Screenings in abgelegenen Gebieten, einschließlich Ländern mit niedrigem und mittlerem Einkommen, und die Beurteilung am Krankenbett mittels tragbarer OCT wird ebenfalls erwartet 1).
Marra KV, Chen JS, Nudleman E, Robbins SL. Review of Retinopathy of Prematurity Management in the Anti-VEGF Era: Evolving Global Paradigms, Persistent Challenges and Our AI-Assisted Future. Clinical & experimental ophthalmology. 2025;53(9):1202-1217. doi:10.1111/ceo.14598. PMID:40908574; PMCID:PMC12747480.
Chiang MF, Quinn GE, Fielder AR, et al. International Classification of Retinopathy of Prematurity, Third Edition. Ophthalmology. 2021 Oct;128(10):e51-e68. doi:10.1016/j.ophtha.2021.05.031. PMID:34247850; PMCID:PMC10979521.
日本眼科学会・日本小児眼科学会. 未熟児網膜症診療ガイドライン(第2版). 2024.
Hamad AE, Moinuddin O, Blair MP, Schechet SA, Shapiro MJ, Quiram PA, et al. Late-Onset Retinal Findings and Complications in Untreated Retinopathy of Prematurity. Ophthalmology. Retina. 2020;4(6):602-612. doi:10.1016/j.oret.2019.12.015. PMID:32059986; PMCID:PMC7282927.
Mintz-Hittner HA, Kennedy KA, Chuang AZ, BEAT-ROP Cooperative Group.. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364(7):603-615. doi:10.1056/nejmoa1007374. PMID:21323540; PMCID:PMC3119530.
Stahl A, Lepore D, Fielder A, Fleck B, Reynolds JD, Chiang MF, et al. Ranibizumab versus laser therapy for the treatment of very low birthweight infants with retinopathy of prematurity (RAINBOW): an open-label randomised controlled trial. Lancet (London, England). 2019;394(10208):1551-1559. doi:10.1016/S0140-6736(19)31344-3. PMID:31522845; PMCID:PMC12316478.
Stahl A, Sukgen EA, Wu WC, Lepore D, Nakanishi H, Mazela J, et al. Effect of Intravitreal Aflibercept vs Laser Photocoagulation on Treatment Success of Retinopathy of Prematurity: The FIREFLEYE Randomized Clinical Trial. JAMA. 2022;328(4):348-359. doi:10.1001/jama.2022.10564. PMID:35881122; PMCID:PMC9327573.
Cryotherapy for Retinopathy of Prematurity Cooperative Group. Multicenter trial of cryotherapy for retinopathy of prematurity: preliminary results. Arch Ophthalmol. 1988;106(4):471-479.
Good WV, Hardy RJ, Dobson V, Palmer EA, Phelps DL, Quintos M, et al. The incidence and course of retinopathy of prematurity: findings from the early treatment for retinopathy of prematurity study. Pediatrics. 2005;116(1):15-23. doi:10.1542/peds.2004-1413. PMID:15995025.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.