Das Retinoblastom ist ein bösartiger Tumor der Netzhaut bei Säuglingen und Kleinkindern. Es entsteht durch die maligne Transformation und Proliferation unreifer Netzhautzellen und gilt als monogene Erkrankung, die durch eine Mutation des RB1-Gens auf dem langen Arm von Chromosom 13 (13q14.2) verursacht wird. Es gibt keine Geschlechterpräferenz, und 95 % der Fälle werden vor dem 5. Lebensjahr diagnostiziert.
Die Inzidenz beträgt 1 pro 15.000–23.000 Geburten, in Japan erkranken jährlich 70–80 Kinder. Das Verhältnis von einseitigen zu beidseitigen Fällen beträgt 3:2, das Durchschnittsalter bei Diagnose liegt bei einseitigen Fällen bei 21 Monaten und bei beidseitigen bei 8 Monaten, wobei beidseitige Fälle früher diagnostiziert werden. In Industrieländern liegt die 5-Jahres-Überlebensrate im lokalisierten intraokularen Stadium bei über 95 %.
Das Retinoblastom wird je nach Art der Genmutation in zwei Hauptkategorien eingeteilt.
Klassifikation
Art der Mutation
Krankheitsmerkmale
Genetisches Risiko
Hereditär (Keimbahnmutation)
RB1-Keimbahnmutation
Häufig beidseitig und multiple Tumoren
Vererbung an 50 % der Kinder
Nicht-hereditär (somatische Mutation)
Somatische Mutation in einer Netzhautzelle
Einseitiger und einzelner Tumor
Keine Vererbung an die nächste Generation
Hereditär (Keimbahnmutation): In allen Körperzellen liegt die erste Mutation vor. Bei der zweiten Mutation entsteht Krebs. Häufig beidseitige und multiple Tumoren, Vererbung an jedes zweite Kind (50 %). Risiko für Sekundärtumoren wie Osteosarkom (15,7 % nach 20 Jahren).
Nicht-hereditär (somatische Mutation): In einer Netzhautzelle sind beide Allele des RB1-Gens mutiert. Einseitiger und einzelner Tumor, kein Vererbungsrisiko für die nächste Generation.
Allerdings sind auch bei einseitigen Fällen einige Keimbahn-RB1-Mutationen enthalten. Man sollte die Vererbbarkeit nicht allein aufgrund der Einseitigkeit verneinen, sondern die Familienanamnese, das Erkrankungsalter und die Tumoranzahl auf der Grundlage von genetischer Beratung und genetischer Bewertung interpretieren. 1)
Die Stadieneinteilung steht in direktem Zusammenhang mit der Strategie für die augenerhaltende Behandlung.
Stadium
Läsionszustand
Richtwert für die Augenerhaltungsrate
T1 (frühe intraokuläre Läsion)
Intraokulär begrenzt, keine Progression
Über 90 %
T2 (fortgeschrittene intraokuläre Läsion)
Intraokuläre Progression
Etwa 50 %
T3
Fortgeschrittene Läsion mit extraokulärer Invasion
Etwa 10 %
QIst das Retinoblastom erblich?
A
Etwa 40 % der Fälle sind erblich (Keimbahn-RB1-Mutation) und werden mit einer Wahrscheinlichkeit von 50 % an das Kind weitergegeben. Die restlichen etwa 60 % sind nicht erblich (nur somatische Mutation) und haben kein Risiko für die nächste Generation. Erbliche Formen neigen zu beidseitigem und multifokalem Auftreten. Zum Zeitpunkt der Diagnose werden Gentests und genetische Beratung empfohlen.
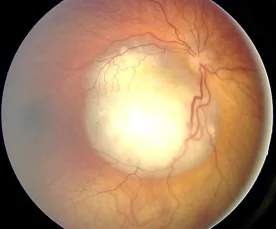
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
Fundusbild eines Retinoblastoms, das als gefäßreiche, weiße, erhabene Läsion beobachtet wird und das typische Erscheinungsbild zeigt, das Leukokorie verursacht. Entspricht den Fundusbefunden und der Leukokorie, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
In vielen Fällen wird der Tumor im Auge groß und zeigt sich als Leukokorie. Wenn er in der Makula auftritt, kann er zu schlechtem Sehen und Schielen führen, was zur Entdeckung beiträgt. Bei älteren Kindern kann eine Verschlechterung des Sehvermögens bemerkt werden; bei Kleinkindern kann das Reiben des schlecht sehenden Auges ein erstes Symptom sein.
Frühsymptome
Leukokorie (weiße Pupille) : häufigstes Erstsymptom. Der Tumor wird im Auge groß und die Pupille erscheint weiß.
Schielen (Strabismus) : verursacht durch schlechtes Sehen aufgrund eines Makulatumors. Das schlecht sehende Auge weicht nach außen ab.
Bewusste Sehverschlechterung : bei älteren Kindern feststellbar.
Augenreiben : bei Kleinkindern mit schlechtem Sehen auf dem betroffenen Auge.
Symptome im fortgeschrittenen Stadium
Hornhauttrübung und erhöhter Augeninnendruck : verursacht durch Kompression der Linse durch den Tumor oder durch ein neovaskuläres Glaukom (NVG) mit erhöhtem Augeninnendruck.
Bindehautrötung und Lidödem : als Entzündungszeichen feststellbar.
Schmerz: Tritt aufgrund von erhöhtem Augeninnendruck oder Tumornekrose auf.
Exophthalmus: Tritt bei extraokulärer Ausbreitung auf.
Es zeigt sich eine gefäßreiche, weißliche, erhabene Läsion; bei Vorliegen von Verkalkungen ist die definitive Diagnose einfach. Häufig begleitet von einer Glaskörperaussaat (Zerfall von Tumorzellen und Verteilung im Glaskörper).
Der rote Reflex ist die Grundlage des Augenscreenings bei Säuglingen. Die Beurteilung ist normal, wenn die Pupillen beider Augen gleich groß sind und ein heller, symmetrischer gelb-oranger Reflex erscheint. Ist der Reflex dunkel oder zu hell, oder besteht ein Seitenunterschied, ist eine weitere Abklärung erforderlich.
QBedeutet eine weiße Pupille immer ein Retinoblastom?
A
Ursachen einer weißen Pupille sind neben dem Retinoblastom zahlreiche andere Erkrankungen wie persistierender hyperplastischer primärer Glaskörper, Frühgeborenenretinopathie, Morbus Coats usw. Bei Vorliegen einer weißen Pupille ist jedoch eine sofortige augenärztliche Untersuchung und der Ausschluss eines Retinoblastoms die höchste Priorität. Eine Verzögerung der Diagnose wirkt sich direkt auf die Prognose aus, daher ist bei Verdacht eine Überweisung noch am selben Tag wünschenswert.
Ursache ist eine Mutation des RB1-Gens auf dem langen Arm von Chromosom 13 (13q14.2). Das RB1-Gen produziert das RB1-Protein (Retinoblastom-Protein), das eine wichtige Rolle bei der Kontrolle der Zellteilung spielt.
Eine Zelle besitzt zwei Genloci; eine Mutation in nur einem Locus erhält die Zellfunktion aufrecht, aber wenn beide Loci mutiert sind, kann die Zelle die Zellteilung nicht mehr kontrollieren und wird bösartig (Zwei-Treffer-Theorie, Knudson-Hypothese).
Hereditäre Form (Keimbahnmutation): Der erste Treffer ist eine Keimbahnmutation (in allen Zellen vorhanden). Tritt in einer somatischen Zelle ein zweiter Treffer auf, kommt es zur Krebsentstehung. Neigung zu bilateralen und multiplen Tumoren.
Nicht-hereditäre Form (somatische Mutation): Sowohl der erste als auch der zweite Treffer treten in derselben somatischen Zelle auf. Sie manifestiert sich als unilateraler, solitärer Tumor.
Die Familienanamnese ist der größte Risikofaktor. Die Risikodefinition gemäß der Empfehlung der AAOOP (American Association of Ophthalmic Oncologists and Pathologists) ist unten aufgeführt1).
Risikoklassifikation
Definition
Risikowert
Hoch
Elternteil mit bilateralem Rb oder Träger einer Keimbahn-RB1-Mutation bei Verwandten ersten oder zweiten Grades
Bei hereditären Fällen ist auf das Risiko von Zweittumoren zu achten. Osteosarkom ist typisch und tritt bei hereditären Fällen innerhalb von 20 Jahren bei 15,7 % auf. Zweittumoren treten häufig nach dem 10. Lebensjahr auf.
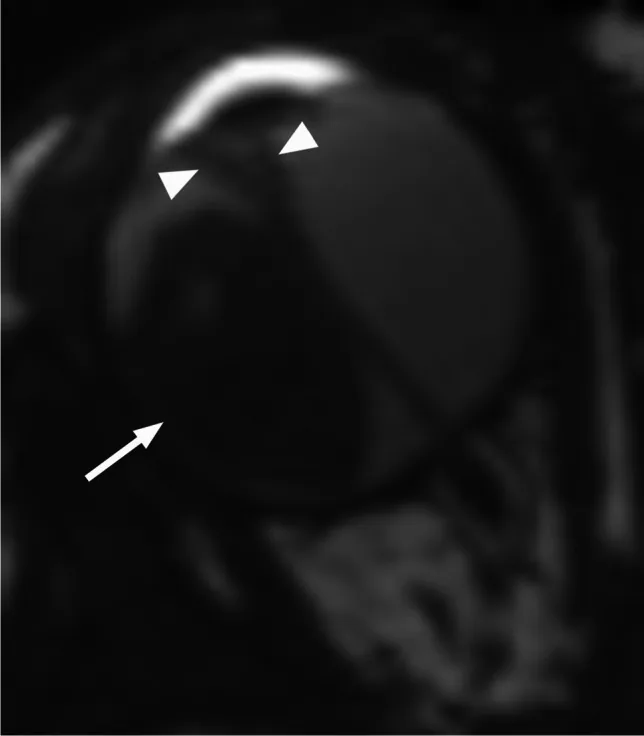
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
Axiale T2-gewichtete MRT zeigt ein exophytisches Retinoblastom (Pfeil) mit einer V-förmigen sekundären Netzhautablösung (Pfeilspitze). Entspricht den in Abschnitt „4. Diagnose und Untersuchungsmethoden“ behandelten exophytischen und endophytischen Wachstumsmustern und MRT-Befunden.
Eine Biopsie intraokularer Tumoren wird nicht durchgeführt. Intraokulare Läsionen können direkt durch transparentes Gewebe beobachtet werden, und die klinische Diagnose ist sehr zuverlässig. Zudem kann eine Biopsie zu einer extraokularen Aussaat von Tumorzellen führen, was ein unvermeidbares Metastasierungsrisiko birgt. Bei augenerhaltender Behandlung wird die Therapie auf Basis der klinischen Diagnose begonnen.
Ultraschall: Bestätigung solider Tumoren und Verkalkungen. Hinweis: Bei Kindern über 5 Jahren ist die Verkalkung gering.
MRT: mittleres Signal in T1, leicht hypointens in T2, Kontrastmittelanreicherung. Unverzichtbar zur Beurteilung von Sehnervinfiltration und extraokularer Ausbreitung.
Kopf-MRT: Bei etwa 3 % der bilateralen Fälle tritt ein trilaterales Retinoblastom (Pinealistumor) auf, daher ist ein Screening obligatorisch.
CT: Hervorragend zur Darstellung von Verkalkungen, aber Strahlenbelastung. Wenn MRT verfügbar, ist CT ergänzend.
Die Differenzialdiagnose von Erkrankungen, die eine Leukokorie verursachen, ist von größter Bedeutung.
Persistierender fetaler Gefäßrest (persistierende hyperplastische primäre Glaskörperpersistenz): Ultraschalluntersuchung zur Bestätigung des Vorhandenseins oder Fehlens eines soliden Tumors
Frühgeborenenretinopathie: Frühgeburtlichkeit und niedriges Geburtsgewicht in der Anamnese sind Hinweise für die Differentialdiagnose
Morbus Coats: Gelb-weißes Exsudat unter der Netzhaut. Das Tumorgefäßmuster ist unterschiedlich
Astrozytäres Hamartom: Differenzierung anhand des Vorhandenseins von Tumorgefäßen, der Lokalisation im OCT und fehlendem Wachstum
Intraokulare Zystizerkose: Selten, kann aber ein Retinoblastom imitieren. Es gibt einen Bericht über einen 4-jährigen Jungen mit Leukokorie, bei dem nach Enukleation aufgrund von Rb-Verdacht die Pathologie eine Zystizerkose ergab3)
Für das Screening von Kindern mit familiärem Rb wird die AAOOP-Empfehlung 2018 international weitgehend herangezogen1).
Risiko
Screening-Intervall
Endalter
Hoch (>7,5%)
Geburt bis 8 Wochen: alle 2–4 Wochen → 8–12 Wochen: monatlich → 1–2 Jahre: alle 2 Monate → 2–3 Jahre: alle 3 Monate → 3–4 Jahre: alle 4 Monate → 4–7 Jahre: alle 6 Monate
7 Jahre (RB1-Mutationsträger lebenslang)
Mittel (1–7,5%)
Geburt bis 3 Monate: monatlich → allmähliche Reduktion
7 Jahre
Niedrig (<1%)
Geburt bis 3 Monate: 1×/Monat → allmähliche Reduktion
7 Jahre
Eine retrospektive nationale Kohortenstudie aus den Niederlanden (1991–2019, 38 familiäre Rb von 332) zeigte, dass alle 28 Patienten mit vollständigem Screening vor dem 1. Lebensjahr diagnostiziert wurden (Median 18 Tage), während bei den 10 Patienten mit unvollständigem Screening die Diagnose erheblich verzögert war (Median 420 Tage, Spanne 59 Tage bis 4,8 Jahre)2). Eine Protokolländerung zur Verkürzung des Screening-Endalters auf 2 Jahre für Niedrigrisikogruppen (<3%) wurde ebenfalls vorgeschlagen2).
Bei familiären Fällen wirkt sich die Fortsetzung des Fundus-Screenings unmittelbar nach der Geburt direkt auf die Prognose aus. Auch in klassischen Registerstudien ist der Diagnosezeitpunkt bei familiären Fällen eng mit der Screening-Häufigkeit verbunden, und derzeit geht man dahin, das Ende des Screenings je nach Vorhandensein einer RB1-Mutation zu individualisieren.1, 2)
QWenn in der Familie ein Retinoblastom aufgetreten ist, wie lange muss das Kind untersucht werden?
A
Die AAOOP empfiehlt regelmäßige Fundusuntersuchungen bis zum Alter von 7 Jahren. Bei vollständigem Screening werden die meisten Fälle vor dem 1. Lebensjahr diagnostiziert. Wenn der Gentest ein RB1-Mutationsrisiko ausschließt, kann das Screening früher beendet werden. Für RB1-Mutationsträger wird nach dem 7. Lebensjahr eine unregelmäßige Nachsorge alle 1–2 Jahre empfohlen.
Bei frühen intraokulären Läsionen mit erhaltener Sehfunktion wird aktiv eine augenerhaltende Behandlung durchgeführt. In fortgeschrittenen intraokulären Stadien ist die Sehfunktion oft nicht zu erwarten, aber auf Wunsch der Familie kann eine erhaltende Behandlung in Betracht gezogen werden. Die Behandlung erfordert ein hohes Maß an Spezialisierung, und eine frühzeitige Überweisung an ein spezialisiertes Zentrum ist wichtig.
Indiziert für Tumore bis zu etwa 3 mm Durchmesser. Durch direkte Bestrahlung mit Infrarotlaser ist eine lokale Kontrolle von etwa 90 % möglich. Bei Tumorbefall der Makula wird eine vorherige systemische Chemotherapie empfohlen, um irreversible Sehschäden zu vermeiden.
② Kryotherapie
Indiziert für Tumore von etwa 3 mm in der Peripherie des Äquators. Die Triple-Freeze-Thaw-Methode (dreimaliges Einfrieren und Auftauen) ist üblich und ermöglicht eine lokale Kontrolle von etwa 90 %, ähnlich wie beim Laser.
③ Brachytherapie
Indiziert für lokalisierte Tumoren mit einer Dicke ≤ 5 mm und einem Durchmesser ≤ 15 mm, die von der Papille entfernt sind. In Japan und Europa wird ¹⁰⁶Ru (Ruthenium-106, β-Strahler) verwendet, in Nordamerika eine ¹²⁵I-Quelle. Die Behandlung besteht in der vorübergehenden Fixierung der Quelle auf der Sklera über dem Tumor, was einen speziellen Behandlungsraum erfordert und die Einrichtungen einschränkt. Eine lokale Kontrolle von 80–90 % ist möglich.
④ Systemische Chemotherapie (VEC-Schema)
Sie ist die Erstlinientherapie für fortgeschrittene intraokulare Tumoren. Eine Dreifach-Kombinationschemotherapie wird häufig eingesetzt, aber die Heilungsrate allein durch diese Behandlung liegt unter 10 %. Nach Tumorverkleinerung wird eine lokale Behandlung (Laser, Kryotherapie, Brachytherapie) zur Konsolidierung durchgeführt.
Medikament
Dosis (basierend auf Körperoberfläche)
Dosis (basierend auf Gewicht für ≤ 36 Monate)
Verabreichungsplan
Vincristin (Oncovin®)
1,5 mg/m²
0,05 mg/kg
Tag 1
Carboplatin (Paraplatin®)
560 mg/m²
18,6 mg/kg
Tag 1
Etoposid (Vepesid®)
150 mg/m²
5 mg/kg
Tag 1, 2
Alle 3–4 Wochen 2–6 Mal wiederholen (alle als intravenöse Infusion).
Direkte Verabreichung eines Medikaments (Melphalan; Alkeran® Injektionslösung) in die Augenarterie mittels Katheter. Durch die lokale Gabe einer hohen Dosis im Auge und die Reduzierung der systemischen Dosis können Nebenwirkungen wie Knochenmarksuppression verringert werden. Es handelt sich um eine nicht von der Krankenkasse abgedeckte, aber in über 20 Ländern durchgeführte experimentelle Behandlung.
Bei Glaskörperaussaat ist die Wirksamkeit von systemischer Chemotherapie und arterieller Injektion begrenzt, daher wird eine intravitreale Injektion von Melphalan (Alkeran® Injektionslösung) ergänzt. Es handelt sich um eine nicht von der Krankenkasse abgedeckte experimentelle Behandlung.
⑦ Externe Strahlentherapie
Fraktionierte Bestrahlung mit 40–46 Gy Röntgenstrahlen. Bis in die 1990er Jahre war sie die Hauptstütze der augenerhaltenden Therapie, aber Deformationen der Augenhöhle und eine Zunahme von Sekundärtumoren wurden offensichtlich, sodass sie heute nur noch eingesetzt wird, wenn andere Behandlungen keine Kontrolle ermöglichen.
Positiver Sehnervenrand und extrasklerale Infiltration sind absolute Indikationen für eine Nachbehandlung, die eine systemische Chemotherapie und Strahlentherapie umfasst. Ausgeprägte Aderhautinfiltration und Sehnerveninfiltration jenseits der Lamina cribrosa werden als relative Risikofaktoren für Metastasen bewertet.
Vorderkammer- oder Irisinfiltration : Risiko extraokulärer Aussaat
Wenn keine Wiederherstellung der Sehfunktion zu erwarten ist : Priorität der Lebensprognose
QBesteht die Möglichkeit, das Auge zu erhalten?
A
Bei frühen intraokularen Läsionen (T1) ist in über 90 % der Fälle ein Augenerhalt möglich. Eine systemische Chemotherapie (VEC-Therapie) verkleinert den Tumor, gefolgt von einer Konsolidierung mittels Laser, Kryotherapie oder Brachytherapie. Bei fortgeschrittenen Fällen (T3) liegt die Augenerhaltungsrate bei etwa 10 %, und eine Enukleation kann erforderlich sein. Die Wahl der Behandlung wird von einem Spezialisten basierend auf dem Stadium und der Sehfunktionsprognose getroffen.
Das RB1-Gen auf dem langen Arm von Chromosom 13 (13q14.2) kodiert für das RB1-Protein (pRb), das eine wichtige Rolle bei der Kontrolle der Zellteilung spielt. pRb bindet an den Transkriptionsfaktor E2F und hemmt den G1/S-Übergang des Zellzyklus, wodurch es als Tumorsuppressorprotein die Zellproliferation reguliert.
Nach der von Knudson vorgeschlagenen Zwei-Treffer-Theorie kommt es zur Malignität, wenn beide Allele des RB1-Gens in einer Zelle inaktiviert werden.
Hereditär: Der erste Treffer (Keimbahnmutation) ist in allen Zellen vorhanden. Wenn in einer Netzhautzelle ein zweiter Treffer (somatische Mutation, LOH usw.) auftritt, entsteht Krebs. Daher treten häufig bilaterale und multiple Tumoren auf, und die Diagnose erfolgt relativ früh.
Nicht-hereditär: Sowohl der erste als auch der zweite Treffer treten als somatische Mutationen in derselben Netzhautzelle auf. Die Wahrscheinlichkeit, dass beide Treffer zufällig in einer Zelle auftreten, ist gering, daher sind die Tumoren oft unilateral und solitär, und die Diagnose erfolgt tendenziell später als bei hereditären Fällen.
Bei hereditären Fällen ist der erste Treffer von RB1 in allen Körperzellen vorhanden. Wenn ein zweiter Treffer in anderen Zellen als der Netzhaut (Knochen, Weichteile usw.) auftritt, entsteht ein sekundärer primärer maligner Tumor. Das Osteosarkom ist am häufigsten und tritt oft nach dem 10. Lebensjahr auf. Bei hereditären Patienten, die eine externe Strahlentherapie erhalten haben, ist das Risiko für Zweitkrebs weiter erhöht, daher wird die externe Strahlentherapie heute nur noch eingeschränkt eingesetzt.
Die IAC (intraarterielle Chemotherapie) injiziert Melphalan direkt in die Augenarterie, wodurch eine hohe Medikamentenkonzentration im Auge bei minimaler systemischer Toxizität erreicht wird. Sie kann auch bei fortgeschrittenen Fällen mit Glaskörperaussaat die Chancen auf Augenerhaltung verbessern. Derzeit laufen Langzeitbewertungen in großen Kohorten.
Die intravitreale Melphalan-Injektion wird zur Behandlung der Glaskörperaussaat durchgeführt, aber die internationale Standardisierung der Dosierungs- und Intervallprotokolle bleibt eine Herausforderung. Mehrere Fallserien berichten über hohe Kontrollraten der Aussaat, und zukünftige prospektive Studien sollen ihre Position etablieren.
In einer niederländischen Kohortenstudie wurde ein klarer Zusammenhang zwischen der Teilnahme am vollständigen Screening und der frühen Diagnose (Median 18 Tage) gezeigt, und die nachteiligen Auswirkungen eines Screening-Abbruchs wurden quantifiziert 2). Darüber hinaus schreitet die Optimierung von Protokollen auf der Grundlage der Risikostratifizierung voran, einschließlich Vorschlägen zur Verkürzung des Screening-Endalters (2 Jahre) für Niedrigrisikogruppen 2). Die weltweite Verbreitung der AAOOP-Empfehlungen und die Standardisierung regionaler Protokolle sind zukünftige Herausforderungen 1).
Obwohl selten, wurden Fälle von Retinoblastom in Verbindung mit angeborenen Hirnfehlbildungen oder Chromosomenanomalien berichtet. Ein Bericht über bilaterales Retinoblastom mit Dandy-Walker-Syndrom unterstreicht die Bedeutung einer systemischen Bewertung einschließlich des neurodevelopmentalen Hintergrunds zusätzlich zu den Augensymptomen. 4)
Mit der Verbreitung der Next-Generation-Sequenzierung (NGS) hat sich die Nachweisempfindlichkeit für Keimbahn-RB1-Mutationen verbessert. Die Möglichkeit der Überwachung mittels Flüssigbiopsie (zirkulierende Tumor-DNA im Blut) wird ebenfalls erforscht, und ihre Anwendung zur prognostischen Stratifizierung ohne invasive Biopsie wird erwartet.
Optimierung der Überwachung von Zweitkrebserkrankungen
Die Standardisierung von Überwachungsprotokollen für Zweitkrebserkrankungen bei Langzeitüberlebenden des hereditären Retinoblastoms ist erforderlich. Insbesondere die Häufigkeit und das Endalter des Multi-Organ-Screenings mittels Ganzkörper-MRT werden durch kontinuierliche Kohortenstudien evidenzbasiert weiterentwickelt.
Die selektive Augenarterieninjektion (IAC) und die intravitreale Chemotherapie (IVitC) etablieren sich international als Behandlungen, die die Augenerhaltungsrate bei fortgeschrittenen Fällen oder Glaskörperaussaat verbessert haben. In Übersichtsarbeiten zur konservativen Behandlung werden sie als Strategien zur Erweiterung der Augenerhaltung ohne Erhöhung des Metastasierungsrisikos positioniert, und die Ergebnisse werden durch den Einführungsstatus in den einzelnen Ländern und die Zentralisierung in spezialisierten Zentren beeinflusst. 5)
Während die 5-Jahres-Überlebensrate in Industrieländern über 95 % liegt, wird berichtet, dass sie in Ländern mit niedrigem und mittlerem Einkommen in Afrika und Asien nur 25–70 % beträgt. Systematische Übersichtsarbeiten und Analysen nach Einkommensniveau der Länder zeigen durchgängig Unterschiede in den Überlebens- und Augenerhaltungsraten, wobei verspätete Diagnose, unzureichender Zugang zur Gesundheitsversorgung und Mangel an spezialisierten Einrichtungen als Hauptfaktoren genannt werden. Die Verbreitung gemeindenaher Screening-Programme, einschließlich der Rotreflex-Methode, ist eine internationale Herausforderung. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.