Das retinale kapilläre Hämangiom (retinal capillary hemangioma) ist ein gutartiger oranger Tumor, der bei jungen Menschen auf der Netzhaut oder der Sehnervpapille auftritt. Aufgrund der pathologischen Ähnlichkeit mit dem zerebralen Hämangioblastom wird es in letzter Zeit auch als retinales Hämangioblastom (retinal hemangioblastoma) bezeichnet. Es kann einzeln/mehrfach, einseitig/beidseitig, sporadisch/syndromal auftreten.
Wenn nur ein sporadisches retinales Hämangioblastom auftritt, spricht man von der Von-Hippel-Krankheit. Bei Vorliegen eines systemischen Tumorsyndroms wird die Diagnose VHL-Krankheit (von-Hippel-Lindau-Krankheit) gestellt. Im Glossar der Japanischen Ophthalmologischen Gesellschaft wird gemäß den VHL-Leitlinien (Ausgabe 2024) der Begriff „retinales Hämangiom“ verwendet, aber in diesem Artikel verwenden wir den gebräuchlichen Begriff „retinales kapilläres Hämangiom“.
Die VHL-Krankheit ist ein autosomal-dominantes erbliches Tumorsyndrom, das durch eine Mutation des Tumorsuppressorgens VHL (3p25-26) verursacht wird. Die Häufigkeit wird mit 1 zu 36.000 angegeben. Bei der VHL-Krankheit treten neben dem retinalen Hämangiom auch Hämangioblastome des Kleinhirns, der Medulla oblongata, der Brücke und des Rückenmarks, Nierenzellkarzinome, Phäochromozytome und Zysten der Bauchorgane (Bauchspeicheldrüse, Nieren, Nebennieren) auf. Die Lebensprognose hängt auch vom Management der systemischen Läsionen ab, daher ist eine multidisziplinäre Zusammenarbeit unerlässlich.
QWann wird eine VHL-Krankheit vermutet?
A
Nach den diagnostischen Kriterien der VHL-Leitlinien (Ausgabe 2024) wird bei positiver Familienanamnese die Diagnose mit einer oder mehreren Läsionen (Hämangioblastom, retinales Hämangiom usw.) gestellt 1). Ohne Familienanamnese wird die Diagnose mit zwei oder mehr Läsionen (einschließlich Hämangioblastom oder retinalem Hämangiom) oder mit Bestätigung einer VHL-Genmutation plus einer Läsion gestellt. Bei jungen Menschen mit sporadischem retinalem Hämangiom, insbesondere unter 10 Jahren, wird später häufig die VHL-Krankheit diagnostiziert, daher sollten eine systemische Untersuchung und ein Gentest aktiv in Betracht gezogen werden.
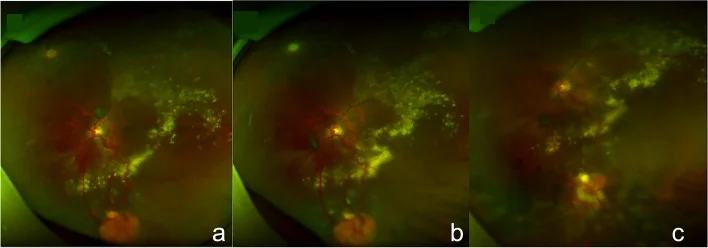
Guo J, Du L, Zhou P, et al. Combined therapy guided by multimodal imaging of fifteen retinal capillary hemangioblastomas in a monocular Von Hippel- Lindau syndrome case report. BMC Ophthalmol. 2022;22(1):205. Figure 4. PMID: 35524216; PMCID: PMC9074324; DOI: 10.1186/s12886-022-02409-8. License: CC BY.
Fundusfotografien, die ein typisches orange-rotes retinales kapilläres Hämangiom in der peripheren Netzhaut mit deutlich erweiterten, geschlängelten versorgenden Gefäßen (zuführende Arterie und abführende Vene) sowie den Verlauf der Tumorrückbildung nach Kryokoagulation zeigen. Sie entsprechen den Fundusbefunden, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
Das retinale Hämangioblastom wird nach Entstehungsweise und Lokalisation wie folgt klassifiziert.
Peripherer Typ
Häufigkeit : Häufigste, typische Form.
Fundusbefund : Bildung eines orangeroten, runden Tumors in der Netzhautperipherie, begleitet von deutlich erweiterten und geschlängelten zuführenden Arterien und abführenden Venen.
Funduskopisch sind solitäre oder multiple runde Netzhauttumoren charakteristisch. Periphere Läsionen sind von erweiterten und geschlängelten zu- und abführenden Gefäßen begleitet und treten in der Regel vor dem 30. Lebensjahr auf. Etwa die Hälfte der Fälle ist bilateral und kann multipel an verschiedenen Stellen des Fundus auftreten.
Der Tumor selbst ist ein Hämangioblastom, das aus Kapillaren und schaumigen Stromazellen besteht und große Mengen an VEGF (vaskulärer endothelialer Wachstumsfaktor) produziert. VEGF führt zu einer exsudativen Netzhautablösung, die eine Sehverschlechterung verursacht.
Im fortgeschrittenen Stadium entwickeln sich folgende schwerwiegende Zustände:
Traktionsnetzhautablösung: Epiretinale Membran und Traktion durch fibröse Proliferation.
Neovaskuläres Glaukom: Endstadium-Komplikation mit erhöhtem Augeninnendruck und Augenschmerzen.
Deutliche Erweiterung der konjunktivalen und episkleralen Gefäße: In extrem fortgeschrittenen Fällen gibt es Berichte über NLP und Augeninnendruck von 45 mmHg im rechten Auge2).
Weitwinkel-Fundusfotografie und OCT-Angiographie (OCTA) sind nützlich für die Beurteilung von Läsionen und die Verlaufskontrolle 1). Die OCT ermöglicht die Beurteilung der Tumormorphologie, des Vorhandenseins von subretinaler Flüssigkeit und eines Makulaödems.
Die Häufigkeit der VHL-Erkrankung beträgt 1 zu 36.000. Die Anzahl der VHL-Patienten in Japan wird auf 600–1.000 geschätzt. Die Inzidenz von retinalen Hämangiomen bei VHL-Patienten liegt bei 40–70 %, das durchschnittliche Erkrankungsalter beträgt 25 Jahre 1).
Altersverteilung : Häufig im Alter von 10–40 Jahren. Ein Auftreten vor dem 10. Lebensjahr tritt in etwa 5 % der Fälle auf.
Bilateralität : Etwa die Hälfte der Fälle ist bilateral.
Multipel : Es können mehrere Tumoren im selben Auge auftreten.
Risiko einer VHL-Progression bei sporadischen Fällen : Bei Kindern unter 10 Jahren, die als sporadisch diagnostiziert wurden, wird bei 45 % später die VHL-Erkrankung diagnostiziert.
Das VHL-Gen folgt einem autosomal-dominanten Erbgang. Eine Keimbahnmutation (erster Hit) liegt auf einem Allel des VHL-Gens vor, und ein zweiter somatischer Hit führt zum Verlust der Tumorsuppressorfunktion (Zwei-Hit-Hypothese).
Als genetischer Risikofaktor benötigen alle Mitglieder von Familien mit einer pathogenen Mutation im VHL-Gen eine Überwachung, und bei Patienten mit retinalem Hämangiom unter 40 Jahren sollte ein VHL-Gentest in Betracht gezogen werden 1).
Bei der Fundusuntersuchung ist die Kombination aus peripheren orange-roten Tumoren und erweiterten, geschlängelten zu- und abführenden Gefäßen charakteristisch. Die Beurteilung erfolgt durch Kombination folgender Untersuchungen:
Weitwinkel-Fundusfotografie : nützlich zur Erfassung des Gesamtbildes peripherer Läsionen.
Fluoreszenzangiographie (FA) : Identifizierung des Tumors und Beurteilung der Aktivität. Ein früher, kräftiger Fluoreszenzaustritt ist diagnostisch wegweisend.
OCT/OCTA : morphologische Beurteilung des Tumors, Quantifizierung von Makulaödem und subretinaler Flüssigkeit.
Ultraschall (B-Modus) : Bestätigung einer soliden Raumforderung und Beurteilung einer Netzhautablösung.
In VHL-Familien beginnt die Fundusuntersuchung unmittelbar nach der Geburt (0 Jahre) und wird mindestens einmal jährlich fortgesetzt 1). Die Kombination aus einer nicht-mydriatischen Funduskamera und Weitwinkel-Fundusfotografie verhindert das Übersehen peripherer Läsionen.
Bei der VHL-Erkrankung können multiple Läsionen auch in anderen Organen als der Netzhaut auftreten, daher ist eine regelmäßige systemische Suche durch ein multidisziplinäres Team unerlässlich 1).
Kopf-MRT : Screening auf Hämangioblastome des Kleinhirns, Hirnstamms und Rückenmarks.
Abdomensonographie, CT/MRT : Suche nach Nierenzellkarzinom, Pankreas-, Nieren- und Nebennierenzysten.
Phäochromozytom-Screening : Messung der Urin-Katecholamin-Metaboliten.
Die oben genannte systemische Untersuchung sollte etwa einmal jährlich fortgesetzt werden.
Der VHL-Gentest ist bei Patienten mit sporadischem retinalem Hämangiom unter 40 Jahren sowie bei Patienten und deren Familien mit Verdacht auf VHL-Krankheit indiziert 1). Es wird empfohlen, ihn parallel zur genetischen Beratung durchzuführen.
Eine Abgrenzung zu folgenden Erkrankungen ist erforderlich.
Morbus Coats: Häufiger bei Jungen. Keine Erweiterung oder Schlängelung der zu- und abführenden Gefäße, aber harte Exsudate und exsudative Netzhautablösung.
Wyburn-Mason-Syndrom (Rankenangiom): Kombination von retinalen und zerebralen arteriovenösen Malformationen.
Aderhaut-Hämangiom: Orangerote Läsion, aber ohne systemische Komplikationen der VHL-Krankheit.
Retinaler vasoproliferativer Tumor: Bevorzugt im unteren peripheren Bereich. Von sekundären Veränderungen abzugrenzen.
QWo finden sich die Fundusbefunde eines retinalen Hämangioms?
A
Der periphere Typ tritt häufig in der Netzhautperipherie auf, daher ist eine detaillierte Fundusuntersuchung unter Mydriasis erforderlich. Kleine Frühformen können wie Mikroaneurysmen aussehen; der Einsatz von Weitwinkel-Fundusfotografie kann helfen, sie nicht zu übersehen. Der papilläre Typ erscheint als peripapilläre Raumforderung, und die Identifikation der zu- und abführenden Gefäße ist oft schwierig.
Die Behandlung wird je nach Lage, Größe und Ausmaß der exsudativen Veränderungen des Hämangioms ausgewählt. Da das Hämangiom bei Größenzunahme therapierefraktär wird, sind Früherkennung und frühzeitige Behandlung wichtig. Eine frühzeitige Behandlung kleiner Läsionen kann die visuelle Prognose verbessern.
Indikation : Erstlinientherapie für den peripheren Typ. Bei Läsionen kleiner als ein Papillendurchmesser kann eine vollständige Heilung erwartet werden.
Methode : Die direkte Koagulation des Hämangioms wird wiederholt, bis ein ausreichender Effekt erzielt wird. Eine frühzeitige Photokoagulation wird auch für kleine Läsionen unter einem Papillendurchmesser empfohlen1).
Grenzen : Bei großen Läsionen über einem Papillendurchmesser sind mehrere Behandlungssitzungen erforderlich.
Kryokoagulation
Indikation : Große Läsionen oder Fälle, bei denen die Laserphotokoagulation schwer zugänglich ist.
Methode : Die Kryokoagulation wird je nach Größe und Vorwölbungsgrad des Hämangioms ausgewählt.
Bei begleitenden exsudativen Veränderungen : Drainage subretinaler Flüssigkeit, Diathermie usw. werden kombiniert, aber die Behandlung ist schwierig.
Traktionsamotio retinae : Bei Fortschreiten der fibrösen Proliferation mit Traktionsamotio wird eine Vitrektomie durchgeführt.
PDT und Anti-VEGF-Therapie : In letzter Zeit häufen sich Fallberichte über die photodynamische Therapie (PDT) und Anti-VEGF-Medikamente (Bevacizumab, Ranibizumab etc.) als Monotherapie oder in Kombination3). Sie werden zur unterstützenden Unterdrückung exsudativer Veränderungen eingesetzt, sind aber nicht erstattungsfähig und erfordern eine Einzelfallentscheidung.
Die Behandlung des papillären Typs ist noch nicht etabliert1). Die Laserphotokoagulation birgt ein hohes Risiko für Schäden am Sehnerv und der Makula, und die Indikationsstellung erfordert besondere Vorsicht. Die folgenden Behandlungen wurden auf Fallberichtsebene beschrieben, aber keine ist als Standardtherapie etabliert.
Intravitreale Injektion von Anti-VEGF-Medikamenten : Eine unterdrückende Wirkung auf exsudative Veränderungen wird erwartet, aber der tumorverkleinernde Effekt ist begrenzt.
Protonenbestrahlung : Konzentrierte Bestrahlung des Tumors5).
Eine sorgfältige Beurteilung des Zeitpunkts des Eingriffs ist erforderlich, und eine multidisziplinäre Konferenz in einer spezialisierten Einrichtung wird empfohlen.
Auch nach der Behandlung treten häufig Rezidive und neue Läsionen auf, daher ist eine lebenslange Nachbeobachtung erforderlich. Nach jeder Behandlung sollte innerhalb von 3–6 Monaten eine Fundusuntersuchung zur Überprüfung der Wirksamkeit und zum Nachweis neuer Läsionen durchgeführt werden. Bei der VHL-Erkrankung treten mehrere Läsionen zeitlich versetzt auf, daher sollte die Untersuchung der gesamten Netzhautperipherie nicht vernachlässigt werden.
Für die Lebensprognose der VHL-Erkrankung ist das Management des Nierenzellkarzinoms und des Hämangioblastoms des zentralen Nervensystems wichtig. In multidisziplinärer Zusammenarbeit mit Neurochirurgie, Urologie, Endokrinologie usw. sollten für jede Organläsion eine angemessene Überwachung und Intervention durchgeführt werden.
6. Pathophysiologie und detaillierte Pathogenesemechanismen
Das VHL-Gen ist ein Tumorsuppressorgen auf Chromosom 3p25-26, das für das pVHL-Protein kodiert, eine Komponente des E3-Ubiquitin-Ligase-Komplexes. Die Hauptfunktion des pVHL-Proteins ist die Ubiquitinierung und der proteasomale Abbau der Hypoxie-induzierten Faktor (HIF) α-Kette.
Unter normalen Bedingungen wird HIFα von pVHL erkannt, ubiquitiniert und schnell abgebaut. Wenn das VHL-Gen inaktiviert wird, geht die pVHL-Funktion verloren und HIFα akkumuliert.
Bei der VHL-Erkrankung führt zusätzlich zur Keimbahnmutation (1. Hit) ein zweiter somatischer Hit (LOH: Verlust der Heterozygotie usw.) zum vollständigen Verlust der VHL-Genfunktion und zur Tumorbildung.
Wenn HIFα im Zellkern akkumuliert, wird die Transkription zahlreicher angiogenese- und zellproliferationsbezogener Gene wie VEGF, PDGF und EPO konstitutiv aktiviert. Bei der VHL-Erkrankung fungiert HIF-2α (EPAS1) als Haupttreiber 1).
Das retinale Angioblastom besteht aus zwei Zelltypen.
Schaumige Stromazellen : Fettreich, produzieren sie zahlreiche Zytokine, darunter VEGF.
Kapillaren : Sekundär induziert durch das von den Stromazellen produzierte VEGF.
Diese Stromazellen bilden den eigentlichen Tumor, und die konstante Produktion von VEGF ist die direkte Ursache für die tumorale Gefäßneubildung und die exsudative Netzhautablösung.
Die Zusammenhänge zwischen VHL-Genmutationstypen und klinischen Phänotypen (Typ 1: nicht-Phäochromozytom-Typ, Typ 2: mit Phäochromozytom assoziierter Typ usw.) sind teilweise bekannt, aber eine detaillierte Klassifikation und Unterschiede in der Häufigkeit von Augenläsionen bedürfen weiterer Analyse.
Belzutifan ist eine niedermolekulare Verbindung, die selektiv HIF-2α hemmt. Im Jahr 2021 wurde es von der FDA (US-amerikanische Food and Drug Administration) für VHL-Patienten mit metastasiertem Nierenzellkarzinom, Hämangioblastom des zentralen Nervensystems oder retinalem Angioblastom zugelassen 7).
Die Ergebnisse der Phase-2-Studie (LITESPARK-004) für retinale Hämangiome sind wie folgt 1).
Ansprechrate : 100 % (12/12 Fälle) zeigten eine Tumorverkleinerung.
Nierenzellkarzinom : Ansprechen bei 49 %.
Hämangioblastom des zentralen Nervensystems : Ansprechen bei 30 %.
Als wichtigste unerwünschte Ereignisse wurden Anämie (ca. 90 %) und Müdigkeit (ca. 66 %) berichtet. Auch in Japan sind klinische Studien geplant, und es wird als vielversprechender Kandidat für die zukünftige Standardtherapie angesehen 1).
In der von Jonasch et al. (2021) berichteten Phase-2-Studie zeigte die orale Gabe von Belzutifan 120 mg einmal täglich bei Patienten mit VHL-Erkrankung eine 100%ige Verkleinerung der retinalen Hämangioblastome 7).
Die photodynamische Therapie (PDT) wurde sowohl für periphere als auch für papilläre Läsionen als geeignet beschrieben.
di Nicola et al. (2022) berichteten über die Wirksamkeit der PDT bei retinalen Hämangioblastomen und zeigten insbesondere ihre Anwendbarkeit bei juxtapapillären Läsionen 4).
Schmidt-Erfurth et al. bewerteten die Anwendbarkeit der PDT bei papillären Läsionen und das Risiko von Komplikationen 6).
Hussain et al. berichteten über die Wirkung der Protonentherapie bei juxtapapillären retinalen kapillären Hämangiomen 5). Als Option für schwierig zu behandelnde papilläre Läsionen häufen sich Fallserien.
Die direkte Tumorverkleinerung ist begrenzt, aber die Berichte über den Einsatz als adjuvante Kontrolle exsudativer Veränderungen nehmen zu 1). Ein Protokoll ist nicht etabliert, und eine individuelle Entscheidung ist erforderlich.
QIst Belzutifan in Japan verfügbar?
A
Stand April 2026 ist Belzutifan in Japan nicht für die allgemeine Anwendung zugelassen. Die FDA hat es 2021 zugelassen, aber in Japan befindet es sich in der Vorbereitungsphase klinischer Studien 1). Bei Patienten mit VHL-Erkrankung, bei denen eine Standardtherapie schwierig ist, sollte die Möglichkeit der Teilnahme an einer klinischen Studie mit dem behandelnden Arzt oder einem spezialisierten Zentrum besprochen werden.
QWas passiert, wenn ein retinales Hämangiom nicht behandelt wird?
A
Wenn das Hämangiom an Größe zunimmt, wird es therapierefraktär und die Sehprognose verschlechtert sich erheblich. Wenn die exsudative Netzhautablösung die Makula erreicht, ist ein irreversibler Sehverlust wahrscheinlich, und ein Fortschreiten zu einer traktiven Netzhautablösung oder einem Neovaskularisationsglaukom kann letztendlich zur Erblindung führen. Bei der VHL-Erkrankung treten lebenslang neue Läsionen auf, daher ist neben der Behandlung eine kontinuierliche Fundusuntersuchung unerlässlich.
Lin H, Lin X. Pronounced conjunctival vascular engorgement in von Hippel-Lindau syndrome: a case report. BMC Ophthalmol. 2020 (症例報告).
Krivosic V, Kamami-Levy C, Jacob J, Richard S, Tadayoni R, Gaudric A.. Laser Photocoagulation for Peripheral Retinal Capillary Hemangioblastoma in von Hippel-Lindau Disease. Ophthalmol Retina. 2017;1(1):59-67. doi:10.1016/j.oret.2016.08.004. PMID:31047395.
Di Nicola M, Williams BK Jr, Hua J, Bekerman VP, Mashayekhi A, Shields JA, Shields CL. Photodynamic Therapy for Retinal Hemangioblastoma: Treatment Outcomes of 17 Consecutive Patients. Ophthalmology. Retina. 2022;6(1):80-88. doi:10.1016/j.oret.2021.04.007. PMID:33892136.
Hussain RN, Jmor F, Damato B, et al. Proton beam radiotherapy for retinal capillary haemangioblastoma. Br J Ophthalmol. 2016;100(3):317-321.
Schmidt-Erfurth UM, Kusserow C, Barbazetto IA, Laqua H.. Benefits and complications of photodynamic therapy of papillary capillary hemangiomas. Ophthalmology. 2002;109(7):1256-1266. doi:10.1016/s0161-6420(02)01059-x. PMID:12093647.
Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ, Thamake S, Park EK, Perini RF, Linehan WM, Srinivasan R, MK-6482-004 Investigators.. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med. 2021;385(22):2036-2046. doi:10.1056/nejmoa2103425. PMID:34818478; PMCID:PMC9275515.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.