Das retinale astrozytäre Hamartom ist ein gutartiger Tumor, der durch eine übermäßige Proliferation von Astrozyten in der Netzhaut entsteht. Ein Hamartom ist eine tumorartige Läsion, die aus normalen reifen Geweben der Region in abnormalen Anteilen besteht. Es besteht kein Risiko einer malignen Entartung.
Diese Erkrankung kann im Rahmen der tuberösen Sklerose (TSC) auftreten oder sporadisch ohne TSC vorkommen. Die tuberöse Sklerose ist eine autosomal-dominante Multisystemerkrankung, die durch Hamartome in verschiedenen Organen gekennzeichnet ist und sich durch Epilepsie aufgrund intrakranieller Läsionen, Talgdrüsenadenome der Haut, renale Angiomyolipome und retinale Hamartome äußert.
Zwei ursächliche Gene sind bekannt: TSC1 (Chromosom 9) und TSC2 (Chromosom 16). TSC1 kodiert für Hamartin, TSC2 für Tuberin, und Funktionsstörungen beider führen zu einer Dysregulation des mTOR-Signalwegs (mechanistic target of rapamycin).
QKann ein retinales astrozytäres Hamartom auch ohne tuberöse Sklerose auftreten?
A
Ja, es gibt sporadische Fälle ohne TSC. Bei sporadischen Fällen ist eine bilaterale Beteiligung selten, und da keine systemischen Komplikationen auftreten, steht die ophthalmologische Betreuung im Vordergrund. Bei TSC-assoziierten Fällen ist hingegen ein multidisziplinäres Management erforderlich, weshalb die Abgrenzung zur TSC bei der Diagnosestellung wichtig ist.
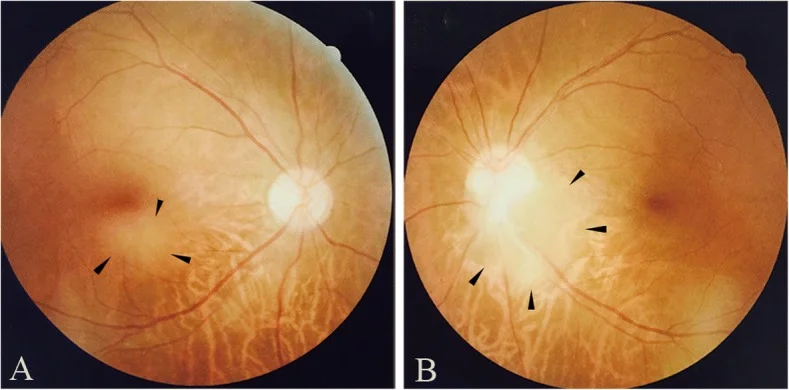
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
Fundusfotografien eines retinalen Astrozytoms im unteren Makulabereich des rechten Auges (a) und am unteren temporalen Rand der Papille des linken Auges (b). Entspricht den Fundusbefunden, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
Das retinale Astrozytom ist oft asymptomatisch und wird zufällig bei einer Screening-Fundusuntersuchung auf tuberöse Sklerose entdeckt. Selten können Sehverschlechterung oder Mouches volantes auftreten. Wenn der Tumor in der Makula lokalisiert ist, sind die Auswirkungen auf das Sehvermögen erheblich.
Der Fundus zeigt charakteristische weißliche erhabene Läsionen. Aufgrund des unterschiedlichen Aussehens werden sie in die folgenden zwei Typen eingeteilt.
Maulbeerartig (Mulberry-Typ)
Aussehen : Kuppelförmige Erhebung mit unebener Oberfläche. Charakteristische Form, die an eine „Maulbeere“ erinnert.
Verkalkung : Häufig mit Verkalkung verbunden. Im Unterschied zur Verkalkung beim Retinoblastom ist die gelbliche Färbung stärker ausgeprägt.
Rand : Aufgrund der langsamen Zellteilung ist die Erhebung gering und der Randanstieg sanft.
Flacher Typ
Aussehen : Weißliche, durchscheinende, flache Läsion, die einem Retinoblastom ähnelt.
Bedeutung der Differenzialdiagnose : Aufgrund der äußerlichen Ähnlichkeit mit einem Retinoblastom ist eine sorgfältige Beurteilung erforderlich.
Verkalkung : Im Vergleich zum Maulbeer-Typ oft ohne Verkalkung.
In der Fluoreszenzangiographie (FAG) werden frühzeitig die feinen Gefäße im Tumor dargestellt. Auch in der Spätphase zeigt sich kein Fluoreszenz-Leck, was ein charakteristischer Befund dieser Erkrankung ist und ein wichtiges Unterscheidungsmerkmal zum Retinoblastom darstellt, das in der Spätphase ein Leck aufweist.
In der optischen Kohärenztomographie (OCT) stellt sich der Tumor als eine hyperreflektive Masse dar, die von den inneren Netzhautschichten aufragt. Charakteristisch sind eine Unordnung der inneren Schichten des Tumors und eine relativ scharfe Abgrenzung zur umgebenden normalen Netzhaut.
Die tuberöse Sklerose (TSC) folgt einem autosomal-dominanten Erbgang. Die Prävalenz liegt bei etwa 1 von 6.000 bis 10.000 Personen. Die Komorbiditätsrate von Netzhaut-Hamartomen bei TSC-Patienten wird mit etwa 50 % angegeben, und sie können beidseitig und multipel auftreten.
Mutationen im TSC1-Gen (Chromosom 9q34) sind mit milderen Symptomen verbunden, während Mutationen im TSC2-Gen (Chromosom 16p13.3) zu schwereren systemischen Läsionen neigen. De-novo-Mutationen sind häufig, daher kann TSC auch ohne Familienanamnese nicht ausgeschlossen werden.
Bei sporadischen Fällen (nicht-TSC) ist der genetische Hintergrund unterschiedlich, und lokalisierte somatische Mutationen können beteiligt sein. Sporadische Fälle sind in der Regel einseitig und einzeln und gehen ohne systemische Komplikationen einher.
Familienanamnese oder gesicherte Diagnose einer tuberösen Sklerose (TSC)
Bekannte genetische Mutationen der TSC (TSC1 oder TSC2)
Vorgeschichte multipler Läsionen des zentralen Nervensystems, der Haut oder der Nieren
QGibt es eine geschlechts- oder altersabhängige Prädisposition?
A
Es wurden keine eindeutigen Geschlechtsunterschiede berichtet. Bei TSC-assoziierten Fällen können Fundusläsionen bereits im frühen Kindesalter festgestellt werden, und eine Fundusuntersuchung wird im Rahmen des pädiatrischen Screenings empfohlen. Sporadische Fälle können auch bei Erwachsenen entdeckt werden, aber alle haben einen gutartigen Verlauf.
Die Diagnose eines retinalen Astrozytenhamartoms erfolgt durch die Kombination charakteristischer Fundusbefunde und systemischer Manifestationen der tuberösen Sklerose.
Differenzialdiagnostische Punkte bei der Fundusuntersuchung:
Weißliche erhabene Läsion mit maulbeerartigem oder flachem Erscheinungsbild
Die wichtigste Differenzialdiagnose ist das Retinoblastom. Bei Kindern mit einer weißlichen erhabenen Netzhautläsion muss zunächst ein Retinoblastom ausgeschlossen werden.
Wird ein retinales Hamartom entdeckt, sollte eine systemische Beurteilung gemäß den TSC-Diagnosekriterien (Northrup-Revision 2012 1)) erfolgen. Zu den Hauptkriterien gehören Adenoma sebaceum (Angiofibrome im Gesicht), Epilepsie, Hirnknoten (Shagreen-Flecken) usw. Für die definitive Diagnose einer TSC ist die Zusammenarbeit mit der Pädiatrie und Neurologie unerlässlich.
Ein retinales astrozytäres Hamartom wächst in der Regel nicht; bei fehlenden Symptomen oder Wachstum ist keine Behandlung erforderlich, und die Überwachung ist die Grundlage. Regelmäßige Fundusuntersuchungen werden durchgeführt, um Wachstum oder Blutungen zu überprüfen.
Wenn die Augenläsion nicht wächst, ist keine aktive Behandlung erforderlich. Bei tuberöser Sklerose sollte die Behandlung systemischer Läsionen (Epilepsie, subependymales Riesenzellastrozytom SEGA, renales Angiomyolipom) gemeinsam mit einem Kinderarzt und Neurologen erfolgen.
mTOR-Inhibitoren (Everolimus) sind für systemische Tumoren bei tuberöser Sklerose (SEGA und renales Angiomyolipom) zugelassen und haben im Rahmen der systemischen TSC-Behandlung eine tumorverkleinernde Wirkung gezeigt2)3). Die Evidenz für eine direkte Wirksamkeit auf retinale Hamartome ist begrenzt, aber es gibt Berichte über Verkleinerung bei Patienten, die im Rahmen einer systemischen TSC-Behandlung behandelt wurden.
QIst eine Behandlung des retinalen astrozytären Hamartoms erforderlich?
A
Bei asymptomatischen und nicht progredienten Fällen ist keine Behandlung erforderlich; die regelmäßige Funduskopie zur Verlaufskontrolle ist die Grundlage. Nur bei wiederholten Blutungen wird eine Vitrektomie oder Netzhautphotokoagulation durchgeführt. Bei tuberöser Sklerose ist neben der ophthalmologischen Betreuung auch das Management der systemischen Erkrankung wichtig.
Die Pathogenese des retinalen astrozytären Hamartoms beruht auf einem Funktionsverlust der Genprodukte von TSC1 (Hamartin) und TSC2 (Tuberin).
Hamartin und Tuberin bilden einen Komplex, der als Tumorsuppressor fungiert, indem er die Aktivität des mTORC1 (mechanistic target of rapamycin complex 1) hemmt. Mutationen in TSC1 oder TSC2 führen zu einem Verlust dieser mTOR-Kontrolle, was zu einer Überaktivierung von mTORC1 führt.
Die Überaktivierung von mTORC1 fördert über die Phosphorylierung von S6-Kinase (S6K) und 4E-BP1 die Zellproliferation, Proteinsynthese und Angiogenese. Dies führt zu einer abnormalen Proliferation von Astrozyten und zur Bildung von Hamartomen (gutartigen Tumoren).
Es wird angenommen, dass die „2-Hit-Hypothese“ an der Tumorentstehung beteiligt ist. Bei Individuen, die in der Keimbahn eine Allelmutation in TSC1 oder TSC2 tragen, führt eine somatische Mutation (zweiter Treffer) in Netzhautzellen zum Funktionsverlust des verbleibenden normalen Allels (LOH: Verlust der Heterozygotie), wodurch die Kontrolle des mTOR-Signalwegs vollständig gestört wird und sich ein Tumor bildet.
Das retinale astrozytäre Hamartom ist ein gutartiger Tumor, der aus einer Proiferation von Astrozyten besteht und nicht maligne entartet. Histologisch sind die abnormal proliferierten Astrozyten dicht angeordnet, teilweise mit Kalziumablagerungen (Verkalkung). Die langsame Zellteilungsrate entspricht dem klinischen Merkmal eines nur langsam wachsenden Tumors.
Everolimus, ein mTORC1-Inhibitor, wird zur Behandlung von tuberöser Sklerose-assoziierten Tumoren eingesetzt. In der EXIST-1-Studie (mit SEGA als Ziel) 2) war der Anteil der Patienten mit einer SEGA-Volumenreduktion von mindestens 50 % in der Everolimus-Gruppe signifikant höher als in der Placebo-Gruppe. Die EXIST-2-Studie (mit renalen Angiomyolipomen als Ziel) 3) berichtete ebenfalls über einen ähnlichen Tumorschrumpfungseffekt.
Hinsichtlich der ophthalmologischen Wirksamkeit von mTOR-Inhibitoren bei TSC-assoziierten retinalen Hamartomen gibt es derzeit nur begrenzte Fallberichte. Bei Patienten, die Everolimus im Rahmen einer systemischen TSC-Behandlung erhielten, wurde eine Verkleinerung retinaler Hamartome beobachtet, jedoch liegen keine Evidenzen aus großen prospektiven Studien vor.
Prognose und Herausforderungen der Langzeitnachsorge
Fundushamartome nehmen in der Regel nicht an Größe zu, können aber in seltenen Fällen wachsen und Blutungen oder Exsudationen verursachen. Die Identifizierung von Risikofaktoren für das Wachstum, die Vorhersage von Sehveränderungen im Langzeitverlauf und die Bestimmung des geeigneten Zeitpunkts für einen Eingriff erfordern eine weitere Fallakkumulation und prospektive Studien.
Pathologie des sporadischen retinalen astrozytären Hamartoms
Der genetische Hintergrund und die molekularen Mechanismen sporadischer Fälle ohne TSC sind noch nicht vollständig aufgeklärt. Es wird vermutet, dass eine lokale Anomalie des mTOR-Signalwegs durch somatische Mutationen beteiligt sein könnte, aber die Aufklärung der genauen Mechanismen ist eine Aufgabe für die zukünftige Forschung.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.