L’amartoma astrocitico retinico è un tumore benigno derivante dalla proliferazione eccessiva degli astrociti della retina. Un amartoma è una lesione tumorale composta da tessuti maturi normalmente presenti in quella sede, ma in proporzioni anomale. Non va incontro a trasformazione maligna.
Questa patologia può essere associata alla sclerosi tuberosa (TSC) o presentarsi in forma sporadica senza TSC. La sclerosi tuberosa è una malattia multisistemica autosomica dominante caratterizzata da amartomi in vari organi, che si manifesta con epilessia da lesioni intracraniche, adenomi sebacei cutanei, angiomiolipomi renali e amartomi retinici.
Sono noti due geni causali: TSC1 (cromosoma 9) e TSC2 (cromosoma 16). TSC1 codifica per l’amartina, TSC2 per la tuberina, e la loro disfunzione porta a una disregolazione della via mTOR (mechanistic target of rapamycin).
QL'amartoma astrocitico retinico può verificarsi in assenza di sclerosi tuberosa?
A
Sì, esistono casi sporadici senza TSC. Nei casi sporadici la bilateralità è rara e, in assenza di complicanze sistemiche, la gestione è prevalentemente oftalmologica. Nei casi associati a TSC, invece, è necessaria una gestione multidisciplinare, quindi è importante differenziare la TSC al momento della diagnosi.
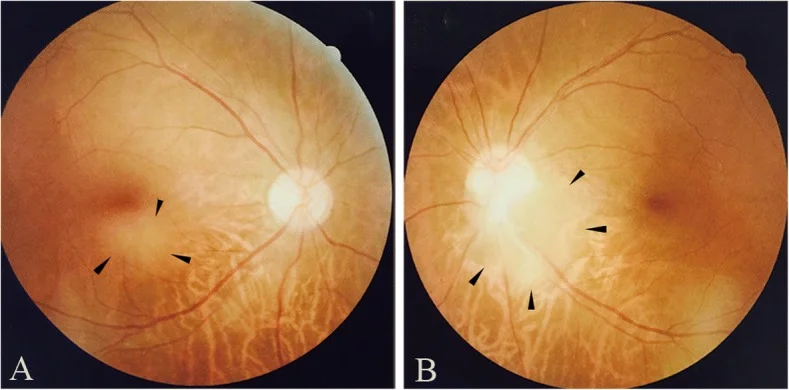
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
Fotografie del fondo oculare che mostrano un astrocitoma retinico nella regione maculare inferiore dell’occhio destro (a) e al margine temporale inferiore del disco ottico dell’occhio sinistro (b). Corrispondono ai reperti del fondo oculare trattati nella sezione «2. Principali sintomi e segni clinici».
L’amartoma astrocitico retinico è spesso asintomatico e viene scoperto incidentalmente durante un esame di screening del fondo oculare per la sclerosi tuberosa. Raramente possono verificarsi diminuzione dell’acuità visiva o miodesopsie. Se il tumore è localizzato nella macula, l’impatto sulla vista è significativo.
Il fondo oculare presenta caratteristiche lesioni biancastre rilevate. In base all’aspetto, vengono classificate nei seguenti due tipi.
Tipo a mora (mulberry)
Aspetto : Rialzo a cupola con superficie irregolare. Forma caratteristica che ricorda una «mora».
Calcificazione : Spesso associata a calcificazione. Rispetto alla calcificazione del retinoblastoma, è caratteristica una tonalità giallastra più marcata.
Margini : Riflettendo una lenta divisione cellulare, il rialzo è lieve e i margini hanno una pendenza dolce.
Tipo piatto
Aspetto : Lesione piatta biancastra traslucida, di aspetto simile al retinoblastoma.
Importanza della diagnosi differenziale : A causa della somiglianza con il retinoblastoma, è necessaria una valutazione attenta.
Calcificazione : Spesso senza calcificazione rispetto al tipo a mora.
All’angiografia con fluoresceina (FAG), i microvasi intratumorali vengono visualizzati precocemente. L’assenza di perdita tardiva di fluoresceina è un reperto caratteristico di questa malattia e costituisce un importante punto di differenziazione dal retinoblastoma, che mostra una perdita tardiva.
Alla tomografia a coerenza ottica (OCT), il tumore appare come una massa iperriflettente che si solleva dagli strati interni della retina. Le caratteristiche includono la disorganizzazione degli strati interni del tumore e un confine relativamente netto con la retina normale circostante.
La sclerosi tuberosa (TSC) segue un modello di ereditarietà autosomica dominante. La prevalenza è di circa 1 persona su 6.000-10.000. Il tasso di comorbilità degli amartomi retinici nei pazienti con TSC è riportato intorno al 50% e possono essere bilaterali e multipli.
Le mutazioni del gene TSC1 (cromosoma 9q34) sono associate a sintomi più lievi, mentre le mutazioni del gene TSC2 (cromosoma 16p13.3) tendono a causare lesioni sistemiche più gravi. Le mutazioni de novo sono frequenti, quindi l’assenza di storia familiare non esclude la TSC.
Nei casi sporadici (non TSC), il background genetico è diverso e possono essere coinvolte mutazioni somatiche localizzate. I casi sporadici sono generalmente unilaterali e singoli, senza complicazioni sistemiche.
Anamnesi di lesioni multiple del sistema nervoso centrale, cutanee o renali
QEsiste una predisposizione legata al sesso o all'età?
A
Non sono state riportate chiare differenze di sesso. Nei casi associati a TSC, le lesioni del fondo oculare possono essere rilevate già nella prima infanzia e si raccomanda l’esame del fondo oculare come parte dello screening pediatrico. I casi sporadici possono essere scoperti anche in età adulta, ma tutti hanno un decorso benigno.
La diagnosi di amartoma astrocitico retinico si basa sulla combinazione di reperti fundoscopici caratteristici e manifestazioni sistemiche della sclerosi tuberosa.
Punti chiave per la diagnosi differenziale all’esame del fondo oculare:
Lesione biancastra rilevata di aspetto a mora o piatto
Assenza di perdita tardiva di fluoresceina all’angiografia
Rilevamento lieve con bordi regolari
Assenza di crescita al follow-up
Tonalità giallastra anche in presenza di calcificazioni
La più importante diagnosi differenziale è il retinoblastoma. Nei bambini con una lesione retinica biancastra rilevata, è necessario prima escludere un retinoblastoma.
Quando viene scoperto un amartoma retinico, è necessario eseguire una valutazione sistemica secondo i criteri diagnostici della TSC (revisione di Northrup 2012 1)). I criteri maggiori includono adenoma sebaceo (angiofibromi facciali), epilessia, noduli cerebrali (macchie di Shagreen), ecc. Per la diagnosi definitiva di TSC è essenziale la collaborazione con i reparti di pediatria e neurologia.
L’amartoma retinico astrocitario di solito non cresce; in assenza di sintomi o crescita, non è necessario alcun trattamento e la sorveglianza è la regola. Si eseguono regolari esami del fondo oculare per verificare l’assenza di crescita o emorragia.
Se la lesione oculare non aumenta, non è necessario un trattamento attivo. Nei casi associati a sclerosi tuberosa, la gestione delle lesioni sistemiche (epilessia, astrocitoma subependimale a cellule giganti SEGA, angiomiolipoma renale) deve essere effettuata in collaborazione con un pediatra e un neurologo.
Inibitori di mTOR (everolimus) sono approvati per i tumori sistemici associati alla sclerosi tuberosa (SEGA e angiomiolipoma renale) e hanno mostrato un effetto di riduzione tumorale nell’ambito del trattamento sistemico della TSC2)3). Le evidenze di efficacia diretta sugli amartomi retinici sono limitate, ma sono stati riportati casi di riduzione in pazienti trattati per TSC sistemica.
QÈ necessario il trattamento dell'amartoma retinico a cellule astrocitarie?
A
In caso di assenza di sintomi e progressione, non è necessario alcun trattamento; il follow-up regolare mediante esame del fondo oculare è la regola. Solo in caso di emorragie ricorrenti si esegue vitrectomia o fotocoagulazione retinica. Nei casi associati a sclerosi tuberosa, la gestione della malattia sistemica è importante in parallelo al follow-up oftalmologico.
La patogenesi dell’amartoma retinico a cellule astrocitarie è dovuta alla perdita di funzione dei prodotti dei geni TSC1 (amartina) e TSC2 (tuberina).
L’amartina e la tuberina formano un complesso che agisce come soppressore tumorale inibendo l’attività di mTORC1 (mechanistic target of rapamycin complex 1). Le mutazioni in TSC1 o TSC2 portano alla perdita di questo controllo mTOR, causando un’iperattivazione di mTORC1.
L’iperattivazione di mTORC1 promuove la proliferazione cellulare, la sintesi proteica e l’angiogenesi attraverso la fosforilazione di S6 chinasi (S6K) e 4E-BP1. Ciò porta a una proliferazione anomala degli astrociti e alla formazione di amartomi (tumori benigni).
Si ritiene che l’ipotesi dei due colpi (2-hit hypothesis) sia coinvolta nello sviluppo del tumore. Negli individui che portano una mutazione di un allele di TSC1 o TSC2 nella linea germinale, una mutazione somatica (secondo colpo) nelle cellule retiniche porta alla perdita di funzione dell’allele normale rimanente (LOH: perdita di eterozigosi), causando una completa disregolazione della via mTOR e la formazione del tumore.
L’amartoma astrocitario retinico è una massa benigna costituita da una proliferazione di cellule di derivazione astrocitaria e non va incontro a trasformazione maligna. Istologicamente, gli astrociti anormalmente proliferati sono disposti densamente, con occasionali depositi di calcio (calcificazione). La bassa velocità di divisione cellulare corrisponde alla caratteristica clinica di una crescita tumorale lenta.
L’everolimus, un inibitore di mTORC1, è utilizzato nel trattamento dei tumori associati alla sclerosi tuberosa. Nello studio EXIST-1 (con target SEGA) 2), la proporzione di pazienti con una riduzione del volume di SEGA di almeno il 50% era significativamente più alta nel gruppo everolimus rispetto al gruppo placebo. Anche lo studio EXIST-2 (con target angiomiolipomi renali) 3) ha riportato un simile effetto di riduzione tumorale.
Per quanto riguarda l’efficacia oftalmica degli inibitori di mTOR sugli amartomi retinici associati a TSC, attualmente esistono solo limitati report di casi. In pazienti trattati con everolimus come parte della terapia sistemica per TSC, è stata osservata una riduzione degli amartomi retinici, ma non sono state stabilite evidenze da studi prospettici su larga scala.
Gli amartomi del fondo oculare di solito non aumentano di dimensioni, ma in rari casi possono crescere e causare emorragie o essudazioni. L’identificazione dei fattori di rischio di crescita, la previsione dei cambiamenti visivi a lungo termine e la determinazione del momento appropriato per un intervento richiedono un ulteriore accumulo di casi e studi prospettici.
Il background genetico e i meccanismi molecolari dei casi sporadici senza TSC non sono ancora completamente chiariti. Si ipotizza che possa essere coinvolta un’anomalia locale della via mTOR dovuta a mutazioni somatiche, ma l’elucidazione dei meccanismi dettagliati è un argomento di ricerca futura.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.