Hamartoma astrositik retina (retinal astrocytic hamartoma) adalah tumor jinak akibat proliferasi berlebihan astrosit (sel glial bintang) di retina. Hamartoma adalah lesi mirip tumor yang terdiri dari komponen jaringan matang yang normalnya ada di lokasi tersebut, namun dengan proporsi abnormal. Tidak menjadi ganas.
Penyakit ini dapat menyertai tuberous sclerosis complex (TSC) atau terjadi secara sporadis tanpa TSC. Tuberous sclerosis complex adalah penyakit multisistem autosomal dominan yang menyebabkan hamartoma di berbagai organ, dengan gejala seperti kejang akibat lesi intrakranial, adenoma sebaceum pada kulit, angiomiolipoma ginjal, hamartoma retina, dan lainnya.
Dua gen penyebab diketahui: TSC1 (kromosom 9) dan TSC2 (kromosom 16). TSC1 mengkode hamartin, TSC2 mengkode tuberin, dan disfungsi keduanya menyebabkan gangguan regulasi jalur mTOR (mechanistic target of rapamycin).
QDapatkah hamartoma astrositik retina terjadi tanpa tuberous sclerosis?
A
Ya, ada kasus sporadis tanpa TSC. Pada kasus sporadis, keterlibatan bilateral jarang, dan tidak ada komplikasi sistemik, sehingga tata laksana oftalmologis menjadi utama. Sementara pada kasus dengan TSC, diperlukan tata laksana multiorgan, sehingga penting untuk membedakan TSC saat diagnosis.
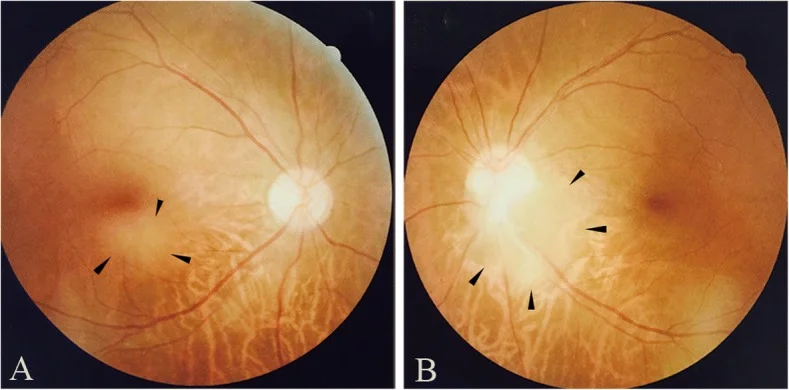
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
Foto fundus yang menunjukkan astrositoma retina di daerah sub-makula mata kanan (a) dan di tepi inferotemporal diskus optikus mata kiri (b). Sesuai dengan temuan fundus yang dibahas di bagian “2. Gejala utama dan temuan klinis”.
Astrositoma retina seringkali asimtomatik, ditemukan secara tidak sengaja pada pemeriksaan fundus skrining tuberous sclerosis. Jarang terjadi penurunan visus atau floaters. Jika tumor berada di makula, dampaknya terhadap penglihatan signifikan.
Pada angiografi fluorescein (FAG), pembuluh darah halus di dalam tumor terlihat pada fase awal. Pada fase akhir, tidak terlihat kebocoran fluorescein, yang merupakan temuan khas penyakit ini, dan menjadi titik diferensiasi penting dari retinoblastoma yang menunjukkan kebocoran pada fase akhir.
Pada optical coherence tomography (OCT), tumor tampak sebagai massa hiperreflektif yang menonjol dari lapisan dalam retina. Ditandai dengan gangguan struktur internal tumor dan batas yang relatif jelas dengan retina normal di sekitarnya.
Tuberous sclerosis complex (TSC) adalah kelainan autosomal dominan. Prevalensinya sekitar 1 dari 6.000–10.000 orang. Insiden hamartoma retina pada pasien TSC dilaporkan sekitar 50%, dan dapat terjadi bilateral atau multipel.
Mutasi gen TSC1 (kromosom 9q34) cenderung menyebabkan gejala ringan, sedangkan mutasi TSC2 (kromosom 16p13.3) lebih sering menyebabkan lesi sistemik yang berat. Mutasi de novo juga sering terjadi, sehingga TSC tidak dapat disingkirkan meskipun tidak ada riwayat keluarga.
Pada kasus sporadis (non-TSC), latar belakang genetik berbeda, dan mungkin melibatkan mutasi somatik terbatas. Kasus sporadis biasanya unilateral dan soliter, tanpa komplikasi sistemik.
Riwayat lesi multipel pada sistem saraf pusat, kulit, atau ginjal
QApakah ada kecenderungan berdasarkan jenis kelamin atau usia?
A
Tidak ada perbedaan jenis kelamin yang jelas dilaporkan. Pada kasus terkait TSC, lesi fundus dapat terdeteksi sejak awal setelah lahir, dan pemeriksaan fundus direkomendasikan sebagai bagian dari skrining pediatrik. Kasus sporadis juga dapat ditemukan pada orang dewasa, namun semuanya memiliki perjalanan jinak.
Penyakit yang paling penting untuk dibedakan adalah retinoblastoma. Jika ditemukan lesi retina putih menonjol pada anak, retinoblastoma harus disingkirkan terlebih dahulu.
Jika ditemukan hamartoma retina, dilakukan evaluasi sistemik berdasarkan kriteria diagnostik TSC (revisi Northrup 2012 1)). Kriteria mayor meliputi adenoma sebaceum (angiofibroma wajah), epilepsi, dan nodul serebral (Shagreen patch). Kolaborasi dengan dokter anak dan neurologi sangat penting untuk diagnosis definitif TSC.
Hamartoma astrositik retina biasanya tidak membesar; jika asimtomatik dan tidak membesar, tidak diperlukan pengobatan dan observasi rutin adalah dasar. Dilakukan pemeriksaan fundus berkala untuk memantau pertumbuhan atau perdarahan.
Jika lesi mata tidak membesar, tidak diperlukan terapi aktif. Pada kasus yang menyertai sklerosis tuberosa, manajemen lesi sistemik (misalnya epilepsi, astrositoma subependimal sel raksasa SEGA, angiomiolipoma ginjal) harus dilakukan bersama dengan dokter anak dan dokter saraf.
Inhibitor mTOR (everolimus) memiliki indikasi asuransi untuk tumor sistemik yang menyertai sklerosis tuberosa (seperti SEGA dan angiomiolipoma ginjal), dan telah dilaporkan efek pengecilan tumor sebagai bagian dari terapi TSC sistemik2)3). Bukti efektivitas langsung pada hamartoma retina terbatas, namun terdapat laporan pengecilan pada kasus yang diberikan dalam konteks terapi TSC sistemik.
Jika tidak bergejala dan tidak membesar, pengobatan tidak diperlukan dan observasi berkala dengan pemeriksaan fundus adalah dasar. Vitrektomi atau fotokoagulasi retina hanya dilakukan pada kasus perdarahan berulang. Pada kasus yang menyertai sklerosis tuberosa, manajemen penyakit sistemik penting dilakukan bersamaan dengan manajemen oftalmologi.
Patogenesis astrositoma hamartoma retina disebabkan oleh hilangnya fungsi produk gen TSC1 (hamartin) dan TSC2 (tuberin).
Hamartin dan tuberin membentuk kompleks yang berfungsi sebagai penekan tumor dengan menghambat aktivitas kompleks mTOR1 (mTORC1). Mutasi pada TSC1 atau TSC2 menyebabkan hilangnya regulasi mTOR ini, sehingga mTORC1 menjadi hiperaktif.
Hiperaktivasi mTORC1, melalui fosforilasi S6 kinase (S6K) dan 4E-BP1, mendorong proliferasi sel, sintesis protein, dan angiogenesis. Akibatnya, astrosit berproliferasi secara abnormal membentuk hamartoma (massa jinak).
Diperkirakan bahwa “hipotesis 2-hit” terlibat dalam perkembangan tumor. Pada individu dengan mutasi alel pada salah satu TSC1 atau TSC2 di garis germinal, terjadinya mutasi somatik (hit kedua) pada sel retina menyebabkan hilangnya fungsi alel normal yang tersisa (LOH: loss of heterozygosity), sehingga regulasi jalur mTOR runtuh sepenuhnya dan tumor terbentuk.
Hamartoma astrosit retina adalah massa jinak yang terdiri dari proliferasi yang berasal dari astrosit, dan tidak mengalami transformasi ganas. Secara histologis, astrosit yang berproliferasi abnormal tersusun rapat, dengan beberapa area mengandung deposit kalsium (kalsifikasi). Kecepatan pembelahan sel yang lambat sesuai dengan karakteristik klinis tumor yang tumbuh lambat.
Everolimus digunakan sebagai inhibitor mTORC1 dalam pengobatan tumor terkait sklerosis tuberosa. Dalam uji coba EXIST-1 (yang menargetkan SEGA) 2), proporsi penurunan volume SEGA sebesar 50% atau lebih secara signifikan lebih tinggi pada kelompok everolimus dibandingkan dengan kelompok plasebo. Efek pengecilan tumor serupa juga dilaporkan dalam uji coba EXIST-2 (yang menargetkan angiomyolipoma ginjal) 3).
Mengenai efektivitas oftalmik inhibitor mTOR pada hamartoma retina terkait TSC, saat ini hanya terdapat laporan kasus terbatas. Ada laporan bahwa pada pasien yang menerima everolimus sebagai bagian dari pengobatan TSC sistemik, terjadi pengecilan hamartoma retina, namun bukti dari uji coba prospektif skala besar belum terbukti.
Prognosis dan tantangan tindak lanjut jangka panjang
Hamartoma retina biasanya tidak membesar, namun dalam kasus yang jarang dapat membesar dan menyebabkan perdarahan atau eksudasi. Diperlukan lebih banyak akumulasi kasus dan studi prospektif untuk mengidentifikasi faktor risiko pembesaran, memprediksi perubahan penglihatan jangka panjang, dan menentukan waktu intervensi yang tepat.
Latar belakang genetik dan mekanisme molekuler kasus sporadik tanpa TSC belum sepenuhnya dipahami. Kemungkinan keterlibatan abnormalitas lokal jalur mTOR akibat mutasi somatik telah disarankan, namun penjelasan mekanisme rinci merupakan subjek penelitian di masa depan.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.