Retinal astrocytic hamartoma is a benign tumor caused by excessive proliferation of retinal astrocytes. A hamartoma is a tumor-like lesion composed of mature tissue components normally present at that site, but in abnormal proportions. It does not become malignant.
This condition can be associated with tuberous sclerosis complex (TSC) or occur sporadically without TSC. Tuberous sclerosis is an autosomal dominant multisystem disorder that causes hamartomas in various organs, presenting with diverse symptoms such as epilepsy due to intracranial lesions, facial angiofibromas, renal angiomyolipomas, and retinal hamartomas.
Two causative genes are known: TSC1 (chromosome 9) and TSC2 (chromosome 16). TSC1 encodes hamartin and TSC2 encodes tuberin, and dysfunction of both leads to dysregulation of the mTOR (mechanistic target of rapamycin) pathway.
QCan retinal astrocytic hamartoma occur without tuberous sclerosis?
A
Sporadic cases without TSC do exist. In sporadic cases, bilaterality is rare, and since there are no systemic complications, ophthalmologic management is the mainstay. In contrast, TSC-associated cases require management of multiple organs, so differentiating TSC is important for definitive diagnosis.
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
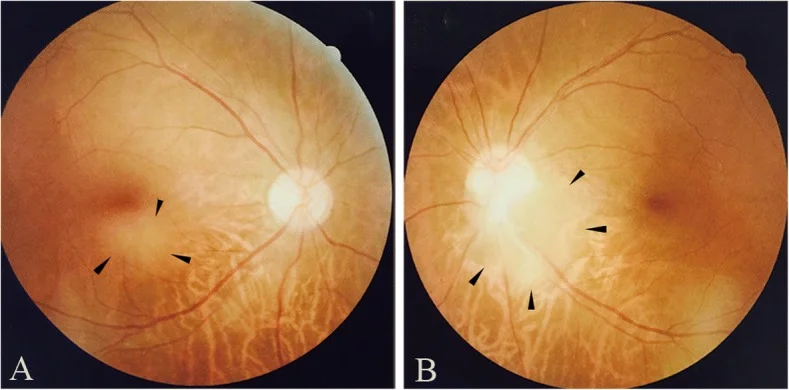
Fundus photographs of retinal astrocytoma observed in the subfoveal region of the right eye (a) and at the inferotemporal margin of the optic disc in the left eye (b). These correspond to the fundus findings discussed in the section “2. Main Symptoms and Clinical Findings.”
Retinal astrocytic hamartoma is often asymptomatic and is discovered incidentally during screening fundus examination for tuberous sclerosis. Rarely, decreased visual acuity or floaters may occur. When the tumor is located in the macula, it has a greater impact on vision.
In fluorescein angiography (FAG), fine intratumoral vessels are visualized in the early phase. The absence of late-phase leakage is a characteristic finding of this disease and an important distinguishing point from retinoblastoma, which shows late-phase leakage.
On optical coherence tomography (OCT), the tumor appears as a hyperreflective mass protruding from the inner retinal layers. Characteristic features include disruption of the internal layered structure and a relatively clear border with the surrounding normal retina.
Tuberous sclerosis complex (TSC) follows an autosomal dominant inheritance pattern. The prevalence is estimated at approximately 1 in 6,000 to 10,000 individuals. The reported rate of retinal hamartoma in TSC patients is about 50%, and it can be bilateral and multifocal.
Mutations in the TSC1 gene (chromosome 9q34) tend to cause milder symptoms, while mutations in the TSC2 gene (chromosome 16p13.3) are more likely to result in severe systemic involvement. De novo mutations are also common, so TSC cannot be ruled out even in the absence of a family history.
In sporadic cases (non-TSC), the genetic background differs, and localized somatic mutations may be involved. Sporadic cases are usually unilateral and solitary, without systemic complications.
History of multiple central nervous system, skin, or renal lesions
QIs there a tendency for onset based on sex or age?
A
No clear sex difference has been reported. In TSC-associated cases, fundus lesions may be detected early after birth, and fundus examination is recommended as part of pediatric screening. Sporadic cases can be discovered even in adults, but all follow a benign course.
The most important differential diagnosis is retinoblastoma. When a white elevated retinal lesion is found in a child, retinoblastoma must first be ruled out.
When a retinal hamartoma is found, systemic evaluation should be performed based on the diagnostic criteria for TSC (Northrup revised version 20121)). Major criteria include facial angiofibroma, epilepsy, and shagreen patch. Collaboration with pediatrics and neurology is essential for a definitive diagnosis of TSC.
Retinal astrocytic hamartoma usually does not enlarge. If asymptomatic and without growth, no treatment is needed and observation is the standard. Regular fundus examinations should be performed to check for growth or hemorrhage.
If the ocular lesion does not enlarge, aggressive treatment is not necessary. In cases associated with tuberous sclerosis, management of systemic lesions such as central nervous system lesions (epilepsy, subependymal giant cell astrocytoma SEGA) and renal angiomyolipoma should be performed together with pediatricians and neurologists.
mTOR inhibitors (everolimus) are covered by insurance for systemic tumors associated with tuberous sclerosis (SEGA and renal angiomyolipoma), and tumor shrinkage effects have been reported as part of systemic TSC treatment2)3). Evidence for direct efficacy against retinal hamartomas is limited, but there are reports of shrinkage in cases where it was administered in the context of systemic TSC treatment.
QIs treatment necessary for retinal astrocytic hamartoma?
A
If asymptomatic and not enlarging, treatment is unnecessary, and regular fundus examination for follow-up is the standard. Vitrectomy or retinal photocoagulation is performed only when bleeding recurs. In cases associated with tuberous sclerosis, management of systemic disease is important in parallel with ophthalmic management.
The pathogenesis of retinal astrocytic hamartoma is due to loss of function of the TSC1 (hamartin) and TSC2 (tuberin) gene products.
Hamartin and tuberin form a complex and function as tumor suppressors that inhibit the activity of mTOR (mechanistic target of rapamycin) complex 1 (mTORC1). When this mTOR regulatory function is lost due to mutations in TSC1 or TSC2, mTORC1 becomes hyperactivated.
Hyperactivation of mTORC1 promotes cell proliferation, protein synthesis, and angiogenesis through phosphorylation of S6 kinase (S6K) and 4E-BP1. As a result, astrocytes proliferate abnormally, forming hamartomas (benign tumors).
The “two-hit hypothesis” is thought to be involved in tumor development. In individuals with a germline mutation in one allele of TSC1 or TSC2, a somatic mutation (second hit) in retinal cells leads to loss of function of the remaining normal allele (LOH: loss of heterozygosity), resulting in complete dysregulation of the mTOR pathway and tumor formation.
Retinal astrocytic hamartoma is a benign tumor composed of proliferating astrocytes, and it does not undergo malignant transformation. Histologically, abnormally proliferating astrocytes are densely arranged, with some areas showing calcium deposits (calcification). The slow cell division rate corresponds to the clinical feature that the tumor grows only slowly.
Everolimus, an mTORC1 inhibitor, is used to treat tuberous sclerosis complex-associated tumors. In the EXIST-1 trial (targeting SEGA) 2), the proportion of patients with a ≥50% reduction in SEGA volume was significantly higher in the everolimus group compared to the placebo group. The EXIST-2 trial (targeting renal angiomyolipoma) 3) also reported similar tumor shrinkage effects.
Regarding the ophthalmic efficacy of mTOR inhibitors for TSC-associated retinal hamartomas, only limited case reports exist at present. There have been reports of retinal hamartoma shrinkage in patients receiving everolimus as part of systemic TSC treatment, but evidence from large-scale prospective trials has not been established.
Retinal hamartomas usually do not enlarge, but in rare cases they may grow and cause hemorrhage or exudation. Identification of risk factors for growth, prediction of visual changes over the long term, and determination of appropriate timing for intervention require further case accumulation and prospective studies.
Pathology of Sporadic Retinal Astrocytic Hamartoma
The genetic background and molecular mechanisms of sporadic cases without TSC are not fully understood. It has been suggested that local mTOR pathway abnormalities due to somatic mutations may be involved, but elucidation of the detailed mechanisms is a topic for future research.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.