早期症狀

視網膜母細胞瘤
1. 什麼是視網膜母細胞瘤?
Section titled “1. 什麼是視網膜母細胞瘤?”視網膜母細胞瘤是嬰幼兒視網膜的惡性腫瘤。它是未成熟視網膜細胞惡變增殖形成腫瘤的結果,被認為是位於13號染色體長臂(13q14.2)的RB1基因突變導致的單基因疾病。無性別差異,95%在5歲前確診。
發生率為1/15,00023,000活產兒,日本每年7080例。單側與雙側比例為3:2,雙側確診較早(平均8個月),單側平均21個月。已開發國家眼內期5年存活率超過95%。
按基因突變分類
Section titled “按基因突變分類”視網膜母細胞瘤根據基因突變的類型大致分為兩類。
| 分類 | 突變類型 | 病理特徵 | 遺傳風險 |
|---|---|---|---|
| 遺傳性(生殖細胞突變) | 生殖細胞系RB1突變 | 常為雙側性和多發性腫瘤 | 子女有50%的遺傳風險 |
| 非遺傳性(體細胞突變) | 單個視網膜細胞的體細胞突變 | 單側性和孤立性腫瘤 | 不會遺傳給下一代 |
遺傳性(生殖細胞突變):身體所有細胞中都存在第一次打擊突變。當發生第二次打擊時,就會致癌。容易引起雙側性和多發性腫瘤,有50%的機率遺傳給子女。存在繼發性癌症(如骨肉瘤,20年風險為15.7%)的風險。
非遺傳性(體細胞突變):視網膜單個細胞中RB1基因的兩個等位基因均發生突變。表現為單側性、孤立性腫瘤,不會遺傳給下一代。
然而,部分單眼病例也包含生殖細胞系RB1突變。不能因為單眼就否定遺傳性,必須在遺傳諮詢和遺傳學評估的前提下,結合家族史、發病年齡和腫瘤數量進行解讀。1)
分期與眼球保留率
Section titled “分期與眼球保留率”分期直接關係到眼球保留治療方案的制定。
| 分期 | 病變狀態 | 眼球保留率參考 |
|---|---|---|
| T1(眼內早期病變) | 侷限於眼內,無進展 | 90%以上 |
| T2(眼內進展期病變) | 眼內進展 | 約50% |
| T3 | 伴有眼外浸潤的進展期病變 | 約10% |
Q
視網膜母細胞瘤會遺傳嗎?
A
約40%為遺傳性(生殖細胞系RB1突變),有50%的機率遺傳給子女。其餘約60%為非遺傳性(僅體細胞突變),無遺傳給下一代的風險。遺傳性病例傾向於雙側和多發性。診斷時建議進行基因檢測和遺傳諮詢。
2. 主要症狀與臨床所見
Section titled “2. 主要症狀與臨床所見”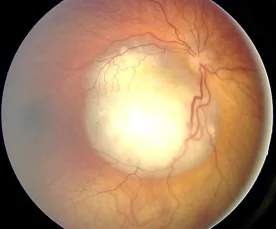
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
多數情況下,腫瘤在眼內增大,表現為白瞳症而被發現。當腫瘤位於黃斑部時,可因視力不良導致斜視而被發現。年長兒童可自覺視力下降,嬰幼兒則可能因視力不良而出現揉眼動作。
可見富含血管的白色隆起病變,若伴隨鈣化則易於確診。常伴有玻璃體播種(腫瘤細胞破碎後散佈至玻璃體)。
紅光反射法(嬰幼兒篩查)
Section titled “紅光反射法(嬰幼兒篩查)”紅光反射法是嬰幼兒眼疾篩查的基礎。判斷標準:雙眼瞳孔大小相等,呈明亮對稱的黃橙色為正常。若反射暗淡或過亮,或左右不對稱,則為異常,需進一步檢查。
Q
白瞳孔一定代表視網膜母細胞瘤嗎?
A
白瞳孔的原因除視網膜母細胞瘤外,還包括永存胎兒血管(永存原始玻璃體增生症)、早產兒視網膜病變、Coats病等多種疾病。但一旦發現白瞳孔,應盡快就診眼科,首先排除視網膜母細胞瘤。診斷延誤直接影響預後,因此若懷疑,建議當日轉診。
3. 原因與風險因素
Section titled “3. 原因與風險因素”RB1基因與二次打擊致癌學說
Section titled “RB1基因與二次打擊致癌學說”病因是13號染色體長臂(13q14.2)上的RB1基因突變。RB1基因編碼RB1蛋白(視網膜母細胞瘤蛋白),該蛋白在調控細胞分裂中起重要作用。
每個細胞內有兩個基因位點,僅一個突變時細胞功能仍可維持,但兩個位點均發生突變時,細胞分裂失控而惡性轉化(二次打擊致癌學說,Knudson假說)。
- 遺傳性(胚系突變):第一次打擊為生殖細胞系突變(存在於所有細胞)。體細胞中發生第二次打擊時致癌。易發生雙側性、多發性腫瘤。
- 非遺傳性(體細胞突變):第一次和第二次打擊均發生於同一個體細胞內。表現為單眼、單發腫瘤。
家族史是最大的風險因子。AAOOP(美國眼科腫瘤與病理學會)建議的風險定義如下所示1)。
| 風險分類 | 定義 | 風險值 |
|---|---|---|
| 高 | 父母患有雙側Rb,或一等或二等親屬為生殖細胞系RB1突變攜帶者 | >7.5% |
| 中 | 父母患有單側Rb | 約7.5% |
| 低 | 其他遠親家族史 | <1% |
二次癌症風險
Section titled “二次癌症風險”遺傳性病例需注意二次癌症的風險。骨肉瘤是典型代表,遺傳性病例中20年內發生率為15.7%。二次癌症常在10歲以後發病。
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”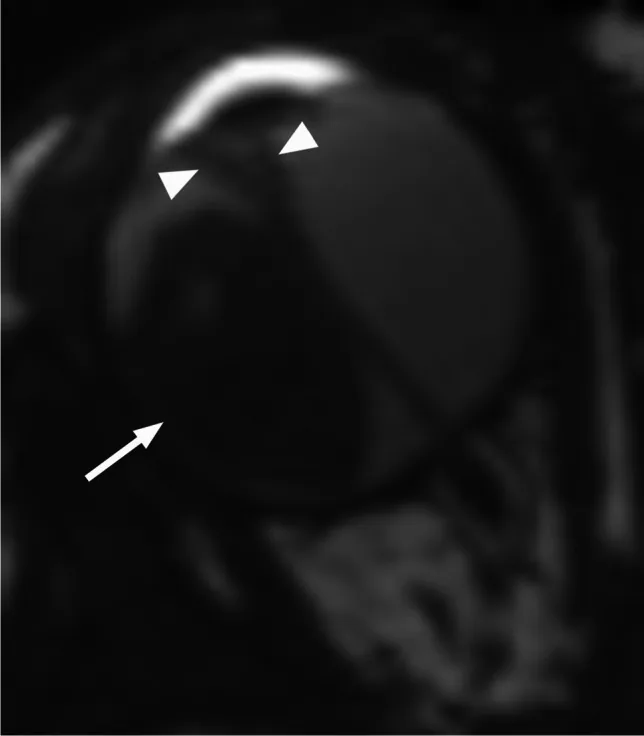
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
軸位T2加權MRI顯示外生型視網膜母細胞瘤(箭頭)伴有V形繼發性視網膜剝離(箭頭頭)。這與本文「4. 診斷與檢查方法」中討論的外生型和內生型生長模式及MRI表現相對應。
診斷基本原則
Section titled “診斷基本原則”不進行眼內腫瘤活檢。眼內病變可透過透明組織直接觀察,臨床診斷準確性高。此外,眼內腫瘤活檢可能導致腫瘤細胞眼外播散,造成不可避免的轉移風險。進行保眼治療時,基於臨床診斷開始治療。
- 眼底檢查(主要):富含血管的白色隆起病變伴鈣化,可臨床確診
- 超音波斷層檢查:確認實質性腫瘤和鈣化。注意5歲以上兒童鈣化較少見
- MRI:T1中等訊號、T2輕度低訊號、有顯影。評估視神經浸潤和眼外擴散必不可少
- 頭部MRI:約3%的雙側病例會發生三側視網膜母細胞瘤(松果體腫瘤),因此必須進行篩查
- CT:顯示鈣化優越,但有輻射。如可行MRI,則CT為輔助地位
最重要的是鑑別引起白瞳症的疾病。
- 胎兒血管殘留(永存原始玻璃體增生症):超音波檢查確認有無實質性腫瘤
- 早產兒視網膜病變:早產、低出生體重病史有助於鑑別
- Coats病:視網膜下黃白色滲出液積聚;腫瘤血管模式不同
- 星形細胞錯構瘤:透過有無腫瘤血管、OCT部位及是否增大進行鑑別
- 眼內囊蟲病:罕見但可模仿Rb。有報導一名4歲男童因白瞳症疑似Rb而行眼球摘除,病理診斷為囊蟲病3)
有家族史時的篩檢
Section titled “有家族史時的篩檢”對於有家族性Rb的兒童篩檢,AAOOP 2018建議在國際上被廣泛參考1)。
| 風險 | 篩檢時間表 | 結束時間 |
|---|---|---|
| 高(>7.5%) | 出生至8週:每2-4週一次 → 8-12週:每月一次 → 1-2歲:每2個月一次 → 2-3歲:每3個月一次 → 3-4歲:每4個月一次 → 4-7歲:每6個月一次 | 7歲(RB1突變攜帶者終身) |
| 中(1-7.5%) | 出生至3個月:每月一次 → 逐漸減少 | 7歲 |
| 低(<1%) | 出生至3個月:每月1次 → 逐漸減少 | 7歲 |
一項荷蘭全國數據的回顧性隊列研究(1991-2019年,332名患者中38名為家族性Rb)顯示,接受完全篩檢的28名患者均在1歲前確診(中位數18天),而不完全篩檢的10名患者確診時間顯著延遲,中位數為420天(59天至4.8歲)2)。此外,有提議將低風險組(<3%)的篩檢終止年齡縮短至2歲的方案修訂2)。
在家族性病例中,出生後立即開始眼底篩檢的持續性直接影響預後。經典登記研究也表明,家族性病例的診斷時機與篩檢頻率密切相關,目前正朝著結合RB1突變有無來個別化終止時間的方向發展。1, 2)
Q
如果家族有視網膜母細胞瘤病史,孩子的篩檢需要持續到什麼時候?
A
AAOOP建議定期進行眼底檢查直至7歲。進行完全篩檢時,絕大多數病例在1歲前確診。如果基因檢測排除RB1突變風險,則可提前終止篩檢。對於RB1突變攜帶者,建議7歲後每1-2年進行不定期追蹤。
5. 標準治療方法
Section titled “5. 標準治療方法”對於眼內早期病變且有望保留視功能的情況,應積極進行保眼治療。在眼內進展期,視功能往往無法保留,但如果家屬希望,可考慮保眼治療。治療需要高度專業性,早期轉診至專科中心至關重要。
① 雷射治療(光凝)
適用於直徑約3mm以內的腫瘤。通過紅外線雷射直接照射,局部控制率可達90%左右。如果腫瘤位於黃斑部,建議先行全身化療以避免不可逆的視功能損害。
② 冷凍凝固
適用於赤道部及周邊直徑約3mm的腫瘤。通常採用三次凍融循環的三重冷凍-解凍法,局部控制率與雷射治療相似,約為90%。
③ 近接放射治療
適應症:腫瘤厚度≤5mm,最大直徑≤15mm,遠離視神經盤的局限性腫瘤。在日本和歐洲使用¹⁰⁶Ru(釕-106,β射線源),在北美使用¹²⁵I放射源。將放射源暫時縫合在腫瘤對應的鞏膜表面。此治療需要特殊治療室,僅在有限機構進行。局部控制率可達80-90%。
④ 全身化學治療(VEC療法)
用於眼內進展期腫瘤的第一線治療。廣泛採用三藥聯合化療,但單獨化療治癒率低於10%。腫瘤縮小後,以局部治療(雷射、冷凍、近接放射治療)進行鞏固。
| 藥物 | 劑量(體表面積基準) | 劑量(≤36個月體重基準) | 給藥時程 |
|---|---|---|---|
| 長春新鹼(Oncovin®) | 1.5 mg/m² | 0.05 mg/kg | 第1天 |
| 卡鉑(Paraplatin®) | 560 mg/m² | 18.6 mg/kg | 第1天 |
| 依托泊苷(Vepesid®) | 150 mg/m² | 5 mg/kg | 第1、2天 |
每3~4週重複2~6次(均為靜脈滴注)。
⑤ 選擇性眼動脈灌注化療(IAC)
通過導管將藥物(美法侖;Alkeran®注射液)直接注入眼動脈。通過向眼部局部給予更多藥物並減少全身藥物量,可以減輕骨髓抑制等副作用。雖然不在健保給付範圍內,但已在全球20多個國家實施,屬於研究性治療。
⑥ 玻璃體注射
對於玻璃體播種,全身化療和動脈灌注的療效有限,因此合併使用美法侖(Alkeran®注射液)的玻璃體注射。這是健保不給付的研究性治療。
⑦ 體外放射治療
分次照射40~46 Gy的X射線。直到1990年代,這曾是保留眼球治療的主要方法,但後來發現會導致眼眶骨變形和繼發性癌症增加,目前僅在其他治療無法控制時使用。
眼球摘除的適應症
Section titled “眼球摘除的適應症”以下情況建議進行眼球摘除。注意盡量長地切除視神經。
- 無法期待視功能時
- 伴有青光眼或蜂窩織炎樣炎症時
- 伴有前房浸潤或虹膜浸潤時
- 懷疑眼外浸潤時
術後輔助治療
Section titled “術後輔助治療”視神經斷端陽性或鞏膜外浸潤是輔助治療的絕對適應症,需進行全身化療和放療。明顯的脈絡膜浸潤或超過篩板的視神經浸潤被視為轉移的相對危險因子。
可保留眼球的條件
需要摘除眼球的條件
Q
有可能保留眼球嗎?
A
對於眼內早期病變(T1),超過90%的病例可以保留眼球。通過全身化療(VEC方案)縮小腫瘤,然後使用雷射、冷凍凝固或近接放射治療進行鞏固。對於進展期病例(T3),眼球保留率約為10%,可能需要摘除眼球。治療方案的選擇由專科醫師根據分期和視功能預期決定。
6. 病理生理學與詳細發病機制
Section titled “6. 病理生理學與詳細發病機制”RB1基因的功能
Section titled “RB1基因的功能”位於13號染色體長臂(13q14.2)的RB1基因編碼RB1蛋白(pRb),該蛋白在調控細胞分裂中起重要作用。pRb通過與E2F轉錄因子結合,抑制細胞週期的G1/S期轉換,從而作為腫瘤抑制蛋白控制細胞增殖。
二次打擊學說(Knudson假說)
Section titled “二次打擊學說(Knudson假說)”Knudson提出的二次打擊學說認為,一個細胞內RB1基因的兩個等位基因均失活才會導致惡性轉化。
- 遺傳性:第一次打擊(生殖細胞系突變)存在於所有細胞中。當視網膜細胞中發生第二次打擊(體細胞突變、LOH等)時,就會發生癌變。因此,容易發生雙側性和多發性腫瘤,並且診斷相對較早。
- 非遺傳性:第一次和第二次打擊均作為體細胞突變發生在同一視網膜細胞內。由於兩次打擊在同一細胞中偶然發生的機率較低,因此腫瘤多為單側性和單發性,診斷時間往往晚於遺傳性病例。
第二原發惡性腫瘤的發生機制
Section titled “第二原發惡性腫瘤的發生機制”在遺傳性病例中,全身細胞均存在RB1的第一次打擊。當視網膜以外的細胞(如骨、軟組織)發生第二次打擊時,就會發生第二原發惡性腫瘤。骨肉瘤最常見,通常在10歲以後發病。接受過外照射放射治療的遺傳性病例,第二原發癌的風險進一步增加,因此目前外照射放射治療的使用受到限制。
7. 最新研究與未來展望
Section titled “7. 最新研究與未來展望”選擇性眼動脈灌注(IAC)適應症的擴大
Section titled “選擇性眼動脈灌注(IAC)適應症的擴大”IAC(眼動脈內化療)通過將美法侖直接注入眼動脈,在最小化全身毒性的同時實現眼內高濃度藥物輸送。即使在伴有玻璃體播散的晚期病例中,也能擴大保留眼球的機會。目前正在進行大規模隊列的長期療效評估。
玻璃體注射的標準化
Section titled “玻璃體注射的標準化”針對玻璃體播散,採用玻璃體內注射美法侖,但國際上的劑量和給藥間隔方案標準化仍是一個挑戰。多個病例系列報告了較高的播散控制率,未來通過前瞻性試驗確立其地位值得期待。
家族性Rb篩查的優化
Section titled “家族性Rb篩查的優化”荷蘭隊列研究明確顯示,完全篩查就診與早期診斷(中位18天)之間存在相關性,並量化了篩查中斷的不利影響2)。此外,針對低風險組縮短篩查終止年齡(至2歲)的提議等,基於風險分層的方案優化正在推進2)。AAOOP建議的全球推廣和區域方案的標準化是未來的挑戰1)。
與先天性腦畸形的合併
Section titled “與先天性腦畸形的合併”雖然罕見,但有病例報告顯示視網膜母細胞瘤與先天性腦畸形或染色體異常並存。伴有Dandy-Walker症候群的雙側視網膜母細胞瘤的報告表明,不僅需要關注眼部症狀,還需要包括神經發育背景在內的全身評估的重要性。4)
基因檢測技術的進步
Section titled “基因檢測技術的進步”隨著次世代定序(NGS)的普及,生殖細胞系RB1突變的檢測靈敏度得到提高。利用液體活檢(循環腫瘤DNA)進行監測的可能性也在研究中,有望應用於避免侵入性活檢的預後分層。
二次癌症監測的優化
Section titled “二次癌症監測的優化”針對遺傳性視網膜母細胞瘤長期存活者的二次癌症監測方案需要標準化。特別是全身MRI多器官篩查的實施頻率和終止時間,正在通過持續的隊列研究累積證據。
IAC和IVitC的國際定位
Section titled “IAC和IVitC的國際定位”選擇性眼動脈灌注(IAC)和玻璃體內化療(IVitC)作為提高晚期病例和玻璃體播散病例眼球保留率的治療方法,正在國際上得到確立。在保守治療的綜述中,它們被定位為在不增加轉移風險的情況下擴大保留眼球機會的策略,各國的實施情況和專業設施的集中化影響療效。5)
發展中國家的治療差距
Section titled “發展中國家的治療差距”已開發國家的5年存活率超過95%,而非洲和亞洲的低收入及中等收入國家報告顯示僅為25%至70%。系統性回顧和按國家收入水準進行的分析一致顯示,存活率和眼球保留率存在差距,主要原因包括延遲診斷、醫療可及性不足以及專業設施缺乏。推廣包括紅反射法在內的社區篩檢計畫是一項國際挑戰。6, 7)
8. 參考文獻
Section titled “8. 參考文獻”- Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
- Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
- Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
- Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
- Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
- Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
- Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.