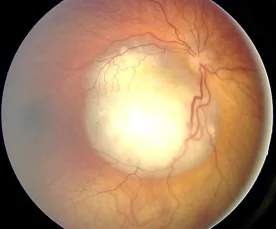
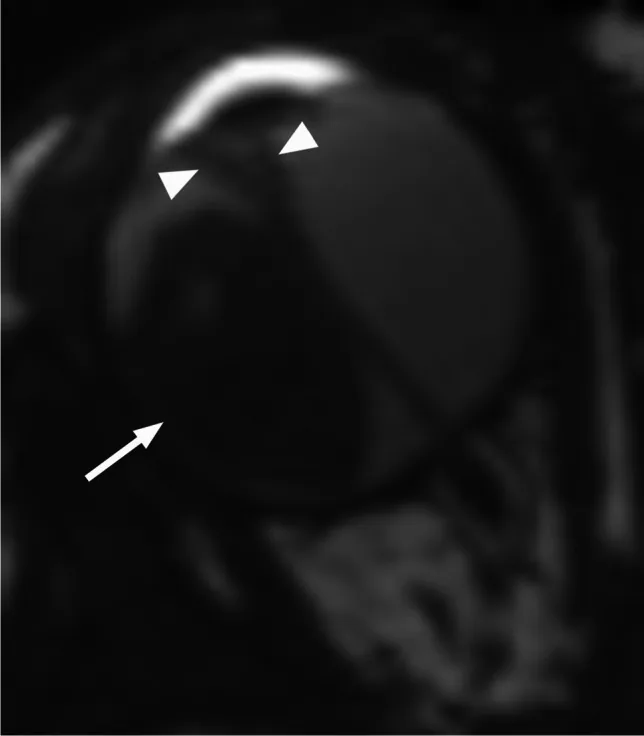
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
คัดลอกข้อความบทความแล้ววางในผู้ช่วย AI ที่คุณต้องการใช้
คัดลอกบทความไปยังคลิปบอร์ดแล้ว
เปิดผู้ช่วย AI ด้านล่าง แล้ววางข้อความที่คัดลอกลงในช่องแชต