
Hemangioma coroidal difuso
1. O que é hemangioma coroidal difuso?
Seção intitulada “1. O que é hemangioma coroidal difuso?”Definição e Conceito
Seção intitulada “Definição e Conceito”Existem dois tipos de hemangioma coroide: localizado (solitário) e difuso. O localizado tem limites nítidos e ocorre esporadicamente, enquanto o difuso é extenso e frequentemente com limites mal definidos. O hemangioma coroide difuso está quase sempre associado à síndrome de Sturge-Weber (angiomatose encefalotrigeminal).
A síndrome de Sturge-Weber é uma das facomatoses (síndromes neurocutâneas), causada pela proliferação ectópica de células da crista neural no período embrionário. Caracteriza-se pela formação de hemangiomas na face, olhos e leptomeninges cerebrais.
Epidemiologia
Seção intitulada “Epidemiologia”A incidência da síndrome de Sturge-Weber é estimada em 1 a cada 20.000 a 50.000 pessoas. Não é hereditária, sendo causada principalmente por uma mutação somática em mosaico no gene GNAQ que ocorre esporadicamente. É uma doença congênita presente desde o nascimento, detectada na infância ou primeira infância. Ocorre no fundo de olho ipsilateral ao hemangioma cutâneo (mancha vinho do Porto) na área de inervação do primeiro e segundo ramos do nervo trigêmeo. Não há dados claros sobre diferenças de sexo ou raça, mas como doença esporádica pode ocorrer em todas as raças.
Comparação com Hemangioma Coroide Localizado
Seção intitulada “Comparação com Hemangioma Coroide Localizado”| Característica | Hemangioma Coroide Difuso | Hemangioma Coroide Localizado (Solitário) |
|---|---|---|
| Síndrome Associada | Sempre com síndrome de Sturge-Weber | Sem doença sistêmica (esporádico) |
| Distribuição no Fundo de Olho | Extensa, limites mal definidos | Localizada, limites relativamente nítidos |
| Coloração | Fundo de olho em ketchup de tomate | Lesão elevada alaranjada |
| Associação com glaucoma | Mais da metade | Raro |
| Época de início | Congênito / infância | Frequentemente na idade adulta |
O hemangioma coroideu difuso ocorre quase sempre associado à síndrome de Sturge-Weber (hemangiomatose encefalofacial). A síndrome de Sturge-Weber apresenta a tríade: mancha vinho do porto facial, hemangioma leptomeníngeo e sintomas oculares (hemangioma coroideu difuso, glaucoma). A incidência é de 1 em 20.000 a 50.000 pessoas, não sendo hereditária. O hemangioma ocorre no fundo de olho ipsilateral à mancha vinho do porto facial.
2. Principais sintomas e achados clínicos
Seção intitulada “2. Principais sintomas e achados clínicos”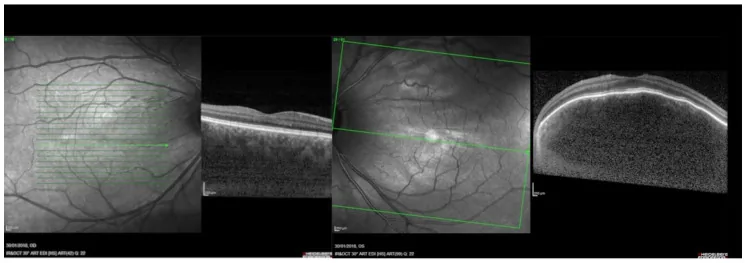
Achados de fundo de olho
Seção intitulada “Achados de fundo de olho”O achado fundoscópico mais característico do hemangioma coroideo difuso é o fundo de olho apresentando uma coloração alaranjada-avermelhada difusa, conhecido como “fundo de olho em ketchup de tomate”. O tumor ocupa a coroide de forma extensa, com bordas mal definidas. Em fotografias de fundo de olho comuns, é possível reconhecer uma diferença acentuada de vermelhidão em comparação com um fundo normal.
Os principais achados fundoscópicos e oftalmológicos são apresentados a seguir.
- Espessamento coroideo difuso: A coroide apresenta-se espessada de forma difusa do polo posterior até a periferia.
- Bordas mal definidas: Diferentemente do hemangioma localizado, as margens do tumor são indistintas.
- Descolamento seroso da retina: Principal causa de baixa acuidade visual. Causa defeitos de campo visual e metamorfopsia.
- Hipermetropização: Ocorre uma alteração refrativa no eixo ocular devido à elevação da coroide.
- Lesão elevada alaranjada: Percebida como uma elevação alaranjada no polo posterior.
Angiografia fluoresceínica e achados de imagem
Seção intitulada “Angiografia fluoresceínica e achados de imagem”Os achados característicos em vários exames de imagem são apresentados a seguir.
- FA (Angiografia fluoresceínica): Hiperfluorescência reticular dentro do tumor desde a fase inicial, com aumento do acúmulo de corante na fase tardia (padrão difuso).
- ICGA (Angiografia com verde de indocianina): Superior à FA na visualização dos vasos coroideos, permitindo uma compreensão clara da estrutura vascular intratumoral.
- OCT: Observado como uma elevação localizada extensa da coroide no polo posterior. Também útil para avaliação de descolamento seroso da retina.
- Ultrassonografia: Visualizada como uma lesão elevada sólida. O espessamento difuso da coroide pode ser confirmado pelo B-scan.
- TC: Visualizada como uma lesão sólida, semelhante à ultrassonografia.
Complicação de glaucoma
Seção intitulada “Complicação de glaucoma”Na síndrome de Sturge-Weber, o glaucoma está presente em mais da metade dos casos. Quanto à época de início, observa-se aproximadamente metade dos casos com início na lactância (tipo congênito) e metade com início na infância (tipo adquirido).
- Tipo de início na lactância: Apresenta características de glaucoma congênito, como aumento do diâmetro da córnea, buftalmia (aumento da córnea) e estrias de Haab. Frequentemente requer cirurgia.
- Tipo de início na infância: A pressão intraocular pode se estabilizar na faixa normal temporariamente, e depois ocorrer elevação da pressão intraocular durante o acompanhamento.
- Hemangioma epiescleral: Alguns casos também apresentam hemangioma na epiesclera do lado afetado.
O glaucoma está presente em mais da metade dos casos de síndrome de Sturge-Weber. O início na lactância e na infância ocorrem em metade dos casos cada, e pode ocorrer elevação tardia da pressão intraocular durante o acompanhamento. Se o glaucoma não for adequadamente manejado, pode levar a danos no campo visual e diminuição da visão, portanto, a medição regular da pressão intraocular e a observação do fundo de olho a longo prazo são essenciais. Especialmente na lactância, buftalmia e opacidade corneana requerem intervenção urgente.
3. Causas e Fatores de Risco
Seção intitulada “3. Causas e Fatores de Risco”Mecanismo de Ocorrência
Seção intitulada “Mecanismo de Ocorrência”A síndrome de Sturge-Weber e o hemangioma coroideo difuso ocorrem devido à proliferação ectópica de células da crista neural durante o período embrionário. Forma-se um hamartoma vascular da coroide, que substitui amplamente a estrutura coroidea normal por tecido vascular.
Em nível genético, a mutação no gene GNAQ (proteína de ligação a nucleotídeos guanina Gqα) (c.548G>A, p.Arg183Gln) é detectada em muitos casos. Essa mutação ocorre como uma mutação mosaico somática, portanto não é hereditária e, em princípio, não é transmitida de pais para filhos. Acredita-se que a mutação GNAQ reduza a atividade GTPase e ative constitutivamente a sinalização de formação vascular.
Principais Fatores de Risco e Características
Seção intitulada “Principais Fatores de Risco e Características”- Mutação mosaico somática no GNAQ: A mais importante como mutação causadora. Não é mutação germinativa, não hereditária.
- Doença congênita: Presente desde o nascimento, portanto mutações incidentais durante o período embrionário são a causa principal, não o histórico genético.
- Fatores ambientais: Não há fatores de risco ambientais estabelecidos.
- Histórico familiar: Em princípio, não é hereditário, portanto geralmente não se observa acúmulo familiar.
Mecanismo de desenvolvimento do glaucoma
Seção intitulada “Mecanismo de desenvolvimento do glaucoma”Dois mecanismos principais estão envolvidos no desenvolvimento do glaucoma na síndrome de Sturge-Weber.
- Aumento da pressão venosa epiescleral: O angioma na epiesclera aumenta a pressão venosa, elevando a resistência ao fluxo de saída do humor aquoso. Mecanismo mais comum no glaucoma que surge após a infância.
- Disgenesia angular: Anomalia no desenvolvimento do ângulo da câmara anterior prejudica o fluxo de saída do humor aquoso. Frequentemente associado ao glaucoma de início na infância.
4. Diagnóstico e métodos de exame
Seção intitulada “4. Diagnóstico e métodos de exame”Abordagem diagnóstica
Seção intitulada “Abordagem diagnóstica”A combinação de mancha vinho do Porto (angioma cutâneo na área de inervação do nervo trigêmeo facial) e alterações fundoscópicas ipsilaterais apoia fortemente o diagnóstico de angioma coroide difuso. Os seguintes exames são usados para diagnóstico oftalmológico.
Exame de fundo de olho e imagem:
- Fotografia de fundo (incluindo fotografia de campo amplo): para registro e acompanhamento do fundo em molho de tomate.
- FA/ICGA: para confirmar hiperfluorescência reticular precoce (padrão difuso).
- OCT: para avaliar espessamento coroidal extenso e descolamento seroso da retina.
- Ultrassonografia (A-scan e B-scan): para confirmar lesão sólida e medir espessura.
Investigação sistêmica:
- RM de crânio: angioma leptomeníngeo e calcificações (avaliado por neurologista pediátrico em conjunto com achados oftalmológicos)
- Tonometria: rastreio de glaucoma (sob anestesia geral se necessário)
- Gonioscopia: avaliação de anomalias do ângulo da câmara anterior
Diagnóstico diferencial
Seção intitulada “Diagnóstico diferencial”As doenças que requerem diferenciação do hemangioma coroideu difuso são apresentadas abaixo.
| Doenças diferenciais | Principais diferenças |
|---|---|
| Hemangioma coroideu focal | Bem delimitado, focal, sem síndrome sistêmica |
| Melanoma maligno coroideu (tipo difuso) | Sinais malignos, alterações pigmentares, risco de metástase |
| Uveíte posterior | Sinais inflamatórios, opacidade vítrea |
| Variação normal da coloração do fundo de olho | Sem achados sistêmicos da síndrome de Sturge-Weber |
Pontos importantes no diagnóstico
Seção intitulada “Pontos importantes no diagnóstico”Quando o fundo do olho parece “levemente vermelho”, especialmente em lactentes com mancha vinho do Porto, deve-se suspeitar fortemente desta doença. Em lactentes, o exame pode exigir anestesia geral, sendo importante a cooperação com os departamentos de anestesiologia e pediatria.
5. Tratamento padrão
Seção intitulada “5. Tratamento padrão”Resumo da estratégia de tratamento
Seção intitulada “Resumo da estratégia de tratamento”O tratamento do hemangioma coroideu difuso é realizado de forma gradual, de acordo com a presença de sintomas e o tipo de complicação. O hemangioma coroideu em si geralmente não é removido cirurgicamente. Os principais alvos do tratamento são o descolamento seroso da retina (principal causa de baixa visual) e o glaucoma.
1. Observação
Seção intitulada “1. Observação”Se não houver sintomas e a visão e a pressão intraocular estiverem normais, continue a observação periódica a seguir:
- Medição da pressão intraocular (detecção precoce de glaucoma)
- Fotografia de fundo de olho e OCT para verificar a presença de descolamento seroso da retina
- Exame de visão e refração (avaliação de hipermetropia e ambliopia)
- Observação periódica com FA/OCT conforme necessário
2. Tratamento para descolamento seroso da retina e declínio da função visual
Seção intitulada “2. Tratamento para descolamento seroso da retina e declínio da função visual”Se houver baixa visual ou descolamento seroso da retina, realiza-se tratamento ativo.
- Administração intravenosa de verteporfina (fotossensibilizador) seguida de irradiação com laser de 689 nm
- A eficácia da PDT no hemangioma coroidal foi relatada
- Não coberto pelo seguro (para hemangioma coroidal difuso)
- Pode-se esperar regressão do descolamento seroso da retina e melhora da visão
- A eficácia no hemangioma coroidal focal está estabelecida, mas a aplicação no tipo difuso tem limitações técnicas devido à grande área de irradiação
Radioterapia de Baixa Dose:
- É administrada radiação de baixa dose de cerca de 20 Gy
- Pode ser eficaz. Espera-se regressão do descolamento da retina e melhora da visão
- Frequentemente é usada radiação externa (teleterapia)
A remoção cirúrgica geralmente não é realizada. O hemangioma coroidal difuso se estende amplamente por toda a coroide, tornando a remoção cirúrgica difícil técnica e funcionalmente. Para o descolamento seroso da retina que causa diminuição da visão, são realizados PDT ou radioterapia de baixa dose (cerca de 20 Gy), sendo frequentemente eficazes. Para o glaucoma, a pressão intraocular é controlada com colírios ou cirurgia.
3. Tratamento do Glaucoma
Seção intitulada “3. Tratamento do Glaucoma”Como mais da metade dos casos apresenta glaucoma, seu manejo é um importante desafio terapêutico.
Terapia Medicamentosa (Colírios):
- Medicamentos relacionados à prostaglandina (inibição da produção de humor aquoso e aumento do fluxo de saída)
- Betabloqueadores (inibição da produção de humor aquoso)
- Inibidores da anidrase carbônica (tópicos ou orais)
- No entanto, no glaucoma devido ao aumento da pressão venosa epiescleral, o efeito dos colírios pode ser limitado
Tratamento cirúrgico:
- No início na infância (tipo congênito), tentam-se goniotomia ou trabeculotomia
- No tipo adulto, considera-se cirurgia filtrante (trabeculectomia ou cirurgia de derivação tubular)
- Se a pressão venosa epiescleral estiver elevada, é necessário cuidado no manejo pós-trabeculectomia
4. Manejo sistêmico
Seção intitulada “4. Manejo sistêmico”- Manejo da epilepsia: Controle de crises com medicamentos antiepilépticos (neurologia pediátrica)
- Abordagem do atraso no desenvolvimento mental: Intervenção precoce e estabelecimento de sistemas de apoio
- Cefaleia e hemiparesia: Observação dos sintomas neurológicos e terapia sintomática
- Colaboração multidisciplinar: Coordenação entre oftalmologia, neurologia pediátrica, dermatologia, especialista em epilepsia e equipe de intervenção precoce
6. Fisiopatologia e mecanismo detalhado de ocorrência
Seção intitulada “6. Fisiopatologia e mecanismo detalhado de ocorrência”Natureza essencial como hamartoma vascular
Seção intitulada “Natureza essencial como hamartoma vascular”O hemangioma coroideo difuso é classificado embriologicamente como um hamartoma vascular. Hamartoma é uma lesão benigna semelhante a um tumor, resultante da proliferação anormal de componentes teciduais normalmente presentes no local, e difere de uma neoplasia verdadeira. Nesta doença, elementos vasculares maduros proliferam excessivamente dentro da coroide, substituindo a estrutura capilar coroidea normal e os vasos médios e grandes.
Mutação do Gene GNAQ e Mecanismos Moleculares
Seção intitulada “Mutação do Gene GNAQ e Mecanismos Moleculares”Como base molecular da síndrome de Sturge-Weber, uma mutação somática em mosaico no gene GNAQ (c.548G>A, p.Arg183Gln) foi identificada em muitos casos.
- Função do GNAQ (Gqα): Subunidade Gα envolvida na sinalização a jusante dos receptores acoplados à proteína G (GPCR)
- Efeito da mutação: A mutação Arg183Gln reduz a atividade GTPase, mantendo o GNAQ em estado constitutivamente ativo
- Sinalização a jusante: Ativação de PLC-β via Gq → produção de IP3/DAG → ativação de PKC → ativação constitutiva da cascata MAPK (MEK/ERK)
- Efeito na angiogênese: A produção excessiva de fatores angiogênicos como VEGF é considerada promotora da formação vascular anormal
- Significado da mutação somática em mosaico: Como a mutação ocorre apenas em algumas células no início do desenvolvimento embrionário, o padrão de expressão varia entre os indivíduos, resultando em diversidade de sintomas
Mecanismo Detalhado do Glaucoma
Seção intitulada “Mecanismo Detalhado do Glaucoma”Na síndrome de Sturge-Weber, pelo menos dois mecanismos são considerados para o desenvolvimento do glaucoma.
Mecanismo 1: Aumento da pressão venosa epiescleral O hemangioma sob a epiesclera (abaixo da cápsula de Tenon) eleva a pressão venosa epiescleral, prejudicando o fluxo do humor aquoso do canal de Schlemm e da malha trabecular. Quando a pressão venosa epiescleral excede o valor normal (cerca de 10 mmHg), a pressão intraocular aumenta proporcionalmente. Esse mecanismo é comum no glaucoma adquirido que surge após a infância.
Mecanismo 2: Disgenesia angular Uma anormalidade no desenvolvimento do ângulo da câmara anterior (disgenesia angular) resulta em desenvolvimento incompleto da malha trabecular, tornando a via de drenagem do humor aquoso anatomicamente não funcional. Esse mecanismo está frequentemente associado ao glaucoma congênito que se manifesta na infância.
Mecanismo do Descolamento Seroso da Retina
Seção intitulada “Mecanismo do Descolamento Seroso da Retina”O aumento da permeabilidade vascular no hemangioma coroideo difuso leva ao extravasamento de componentes intravasculares para o espaço extravascular, com acúmulo de líquido no espaço sub-retiniano. Isso causa descolamento seroso da retina, sendo a principal causa de baixa visual e defeitos de campo visual. A terapia fotodinâmica (PDT) ou radioterapia reduz os vasos tumorais, diminuindo a exsudação e permitindo a reaplicação da retina.
Fisiopatologia Extraocular
Seção intitulada “Fisiopatologia Extraocular”- Angioma leptomeníngeo cerebral: O angioma no córtex cerebral causa isquemia local e calcificação, levando a epilepsia, atraso no desenvolvimento mental e hemiparesia.
- Mancha vinho do Porto facial: Presente desde o nascimento como ectasia capilar na pele.
- Calcificação: Calcificação em “trilho de trem” no córtex cerebral abaixo do angioma leptomeníngeo é uma característica de imagem da síndrome de Sturge-Weber.
7. Pesquisas recentes e perspectivas futuras
Seção intitulada “7. Pesquisas recentes e perspectivas futuras”Terapia alvo molecular direcionada à via downstream de GNAQ
Seção intitulada “Terapia alvo molecular direcionada à via downstream de GNAQ”A cascata MAPK (via MEK/ERK) constitutivamente ativada pela mutação GNAQ tem sido amplamente estudada como alvo terapêutico na pesquisa de melanoma uveal. Como a mesma anormalidade molecular está presente na síndrome de Sturge-Weber/hemangioma coroidal difuso, a aplicação de inibidores de MEK (trametinibe, binimetinibe, etc.) e inibidores diretos de GNAQ (YM-254890, etc.) está sendo considerada em nível de pesquisa básica.
Na verdade, a terapia alvo molecular para toda a síndrome de Sturge-Weber com inibidores de mTOR (sirolimo) e terapias visando a via PI3K-AKT-mTOR foi realizada experimentalmente, com relatos de melhora dos sintomas em alguns pacientes. No entanto, estudos em larga escala avaliando diretamente a eficácia no hemangioma coroidal difuso em si são atualmente limitados.
Otimização do tratamento com PDT
Seção intitulada “Otimização do tratamento com PDT”A PDT tem se mostrado eficaz para o descolamento seroso da retina devido ao hemangioma coroidal difuso em vários relatos de casos, mas o protocolo (campo de irradiação, energia, número de sessões) para lesões difusas extensas não foi padronizado. Comparado ao hemangioma coroidal focal, a área de irradiação é maior no tipo difuso, apresentando desafios técnicos. Espera-se o estabelecimento de protocolos por meio de estudos prospectivos multicêntricos no futuro.
Novos desenvolvimentos no manejo do glaucoma
Seção intitulada “Novos desenvolvimentos no manejo do glaucoma”A cirurgia filtrante convencional para glaucoma devido ao aumento da pressão venosa epiescleral apresenta risco de efusão coroidal e glaucoma maligno em condições de hipertensão venosa epiescleral. Pesquisas estão em andamento sobre a eficácia da cirurgia de derivação tubular (válvula de Ahmed, tubo de Baerveldt). A possibilidade de aplicação da trabeculoplastia a laser seletiva (SLT) também está sendo estudada.
Perspectivas do diagnóstico genético de GNAQ
Seção intitulada “Perspectivas do diagnóstico genético de GNAQ”A detecção da mutação GNAQ em biópsia de pele e amostras de sangue está sendo aplicada para o diagnóstico definitivo da síndrome de Sturge-Weber. No futuro, espera-se que o diagnóstico precoce por biópsia líquida (ctDNA) e a melhora na taxa de detecção de mutações permitam um diagnóstico mais preciso.
8. Referências
Seção intitulada “8. Referências”- Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J.. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971-1979. doi:10.1056/nejmoa1213507. PMID:23656586; PMCID:PMC3749068.
- Baselga E, Torrelo A, Mediero IG, et al. Sturge-Weber syndrome: report of 3 cases. Pediatr Dermatol. 2019;36(6):932-934.
- Bhatt A, Bhatt N. Sturge-Weber syndrome: a rare neurocutaneous disorder. J Pediatr Neurosci. 2021;16(1):1-6.
- Zreik O, Elabassy HM, Bakhurji E, Al-Johani SM. Sturge-Weber syndrome: an updated review. Clin Ophthalmol. 2023;17:2369-2381.
- Nassiri N, Rootman DB, Rootman J, Goldberg RA. Orbital and adnexal lymphangiomas: a review of management. Surv Ophthalmol. 2015;60(3):245-257.