Iris
Pupille en trou de serrure : typiquement, le défaut est situé en bas et en dedans, déformant la pupille en forme de trou de serrure.
Localisation inféro-temporale : peut également survenir dans des positions atypiques.

Le colobome (coloboma) tire son origine du mot grec signifiant « défaut » et est une maladie congénitale caractérisée par des défauts tissulaires dans diverses parties de l’œil dus à une fermeture incomplète de la fissure embryonnaire. Il peut survenir au niveau des paupières, de l’iris, du cristallin, du corps ciliaire, de la choroïde, de la rétine et du nerf optique. Le défaut est typiquement situé en position inféro-nasale et est souvent associé à une microphtalmie.
La prévalence est estimée entre 0,5 et 2,2 cas pour 10 000 naissances. Aux États-Unis, elle est d’environ 2,6 cas pour 10 000 naissances 4), tandis qu’en Europe, elle est rapportée entre 4 et 19 cas pour 100 000 naissances 6). Elle représente environ 11 % des cas de cécité infantile, et le taux de diagnostic génétique reste inférieur à 30 % 6). La prévalence du colobome palpébral est de 0,2 à 0,8 cas pour 10 000 naissances. Il représente 0,07 % des malformations oculaires congénitales et 3,2 à 11,2 % chez les enfants malvoyants.
Il existe des colobomes typiques et atypiques. Les colobomes typiques résultent d’une fermeture incomplète de la fente embryonnaire et sont situés dans le quadrant inféro-nasal, tandis que les colobomes atypiques surviennent dans d’autres régions et impliquent des mécanismes de développement différents.
Les codes CIM-10 sont Q10.3 (paupière), Q13.0 (iris), Q12.2 (cristallin), H47.319 (nerf optique) et Q14.8 (choroïde/rétine).
Il existe à la fois des formes sporadiques et héréditaires. Divers modes de transmission ont été rapportés, notamment autosomique dominant, autosomique récessif et lié à l’X. Plusieurs gènes responsables ont été identifiés, tels que PAX2, CHD7 et FZD5, mais le taux de diagnostic génétique est inférieur à 30 % 6). Un conseil génétique est recommandé en cas d’antécédents familiaux.
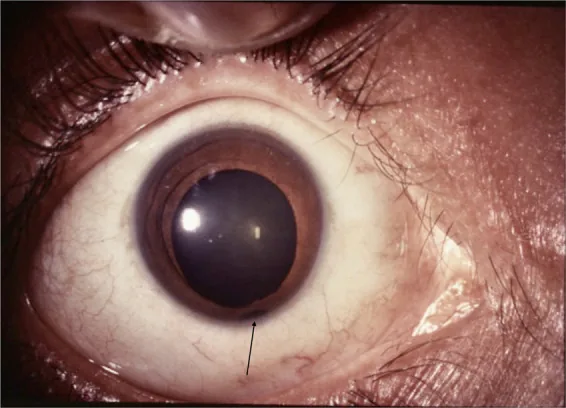
L’acuité visuelle varie considérablement, allant de l’absence de perception lumineuse à une vision normale, selon la localisation et l’étendue du défaut.
Le colobome présente des signes caractéristiques dans chaque partie de l’œil.
Iris
Pupille en trou de serrure : typiquement, le défaut est situé en bas et en dedans, déformant la pupille en forme de trou de serrure.
Localisation inféro-temporale : peut également survenir dans des positions atypiques.
Choroïde et rétine
Lésions blanc-jaunâtre : défaut circulaire à en forme d’éventail, aux bords nets, laissant voir la sclère.
Risque de décollement de la rétine : incidence de 23 à 40 %7). Une surveillance régulière est nécessaire.
Nerf optique et cristallin
Élargissement de l’excavation du nerf optique : unilatéral ou bilatéral, de degré variable.
Aplatissement de l’équateur du cristallin : dû à un défaut de la zonule de Zinn. Observé sous dilatation pupillaire.
Paupière
Défaut interne de la paupière supérieure : perte de tissu sur toute l’épaisseur.
Association à des malformations systémiques : parfois isolé, mais peut être associé à d’autres anomalies.
Le colobome ciliaire isolé est rare ; il est souvent observé en continuité avec un grand colobome choroïdien.
L’acuité visuelle varie de l’absence de perception lumineuse à une vision normale. Si le colobome est limité à l’iris, la vision est souvent préservée. Lorsque la macula ou le nerf optique est impliqué, la vision a tendance à être mauvaise.
La principale cause du colobome est la fermeture incomplète de la fente embryonnaire.
La fente embryonnaire (fente optique) se forme à la 4e semaine de gestation et se termine à la 5e semaine. La fermeture commence à la 6e semaine et s’achève à la 7e semaine. Si ce processus de fermeture est perturbé pour une raison quelconque, un colobome se développe. Le rôle de la vitamine A a également été suggéré.
Plusieurs gènes impliqués dans le développement du colobome ont été identifiés.
| Gène | Maladie/Phénotype associé |
|---|---|
| PAX2 | Syndrome rénal-colobome5) |
| CHD7 | Syndrome CHARGE |
| FZD5 | Colobome oculaire symptomatique + microcornée6) |
| TENM3 | MCOPS15 (microcornée + retard de développement) 8) |
| FAT1 | Colobome + néphropathie 9) |
| YAP1 | Associé au colobome |
| ABCB6 | Associé au colobome |
| SALL2 | Associé au colobome |
Le colobome peut être associé aux syndromes systémiques suivants :
C’est un syndrome de malformations multiples dû à une mutation du gène CHD7. Le nom est formé par les initiales de colobome (C), cardiopathie (H), atrésie des choanes (A), retard de croissance et de développement (R), hypoplasie génitale (G) et anomalies des oreilles (E). Le diagnostic repose sur une combinaison de ces signes.
Des tests génétiques complets comme le séquençage de l’exome entier (WES) sont réalisés, mais le taux de diagnostic reste inférieur à 30 %6).
Le colobome nécessite un diagnostic différentiel selon la localisation, comme suit.
| Localisation | Principaux diagnostics différentiels |
|---|---|
| Paupière | Syndrome de brides amniotiques, traumatisme |
| Iris | Aniridie, déchirure traumatique de l’iris |
| Nerf optique | Syndrome de la fleur de liseron, hypoplasie du nerf optique |
Il n’existe pas de traitement curatif du colobome ; la prise en charge repose sur un traitement symptomatique et la gestion des complications selon la localisation.
Castilla-Martinez et al. (2024) ont réalisé une chirurgie de la cataracte au laser femtoseconde (FLACS) combinée à une pupilloplastie et à la mise en place d’un CTR chez un patient présentant un colobome de l’iris, du cristallin et de la zonule associé à une cataracte. L’acuité visuelle postopératoire s’est améliorée à logMAR 0,24).
Dans le colobome du nerf optique, en raison de l’hypoplasie de la lame criblée, l’artère et la veine centrales de la rétine se divisent déjà en arrière de la papille, et les vaisseaux rétiniens naissent de multiples sites à la périphérie de la papille. Une atrophie choroïdienne et rétinienne due à une fermeture incomplète de la fente embryonnaire est souvent observée sous la papille.
Pour le décollement rhegmatogène de la rétine, une vitrectomie est réalisée. Des techniques chirurgicales telles que la réapplication rétinienne avec colle de fibrine 7) et la photocoagulation endoculaire avec tamponnement au gaz 3) ont été rapportées. En cas de décollement séreux, une régression spontanée est possible, et la stratégie thérapeutique est décidée individuellement.
La cupule optique se forme à partir du neuroectoderme à la 4e semaine de gestation. Une fissure fœtale (fissure optique) apparaît sur la face ventrale de la cupule optique, permettant le passage de l’artère hyaloïde. Cette fissure se complète à la 5e semaine et commence à se fermer à partir de la 6e semaine. La fermeture débute près de l’équateur et progresse vers l’avant (côté irien) et vers l’arrière (côté du nerf optique), pour s’achever à la 7e semaine.
Le processus de fermeture implique la transition épithélio-mésenchymateuse (TEM). Les cellules épithéliales de la rétine neurale au bord de la fissure fœtale dégradent la membrane basale, acquièrent un phénotype mésenchymateux et fusionnent. Une perturbation de ce processus conduit à la formation d’un colobome.
Le gène FZD5 code pour un récepteur de la voie de signalisation Wnt. Les mutations hypofonctionnelles de FZD5 entraînent une anomalie de la signalisation Wnt, provoquant un défaut de fermeture de la fissure fœtale et une microcornée 6).
Les cellules de la crête neurale (CCN) sont également impliquées dans la genèse du colobome. Les CCN se différencient en tissu mésenchymateux autour de la cupule optique et jouent un rôle important dans le processus de fermeture de la fissure fœtale 2). Une perturbation de la migration des CCN entraîne des anomalies du développement de l’iris et de la choroïde.
Cortes-Gonzalez et al. (2024) ont rapporté qu’une mutation homozygote faux-sens de FZD5 (p.M160V) provoque un colobome oculaire symptomatique et une microcornée 6). La transmission est récessive, et l’analyse fonctionnelle a confirmé une altération de l’activation dépendante du ligand de la voie de signalisation Wnt. Le taux de diagnostic génétique du colobome est inférieur à 30 %, et l’identification de nouveaux gènes responsables devrait améliorer le diagnostic.
Zhou et al. (2022) ont rapporté que des mutations hétérozygotes composites du gène TENM3 provoquent le MCOPS15 (microcornée, colobome irido-choroïdien, retard global du développement) 8). TENM3 code une protéine transmembranaire impliquée dans l’adhésion cellulaire et la neurogenèse.
Esmaeilzadeh et al. (2022) ont rapporté l’identification de mutations du gène FAT1 dans une famille iranienne présentant un colobome irien et une néphropathie 9). FAT1 est un membre de la superfamille des cadhérines impliqué dans la polarité cellulaire et la morphogenèse tissulaire.
Hu et al. (2024) ont rapporté l’identification d’une mutation frameshift c.76delG de PAX2 dans une famille atteinte de glomérulosclérose segmentaire et focale (FSGS) 5). Cette observation suggère que le spectre phénotypique du syndrome de colobome rénal est plus large qu’on ne le pensait.
Jain et al. (2024) ont rapporté un cas de décollement de rétine associé à un colobome traité par rétinopexie avec colle de fibrine 7). La technique consiste à appliquer de la colle de fibrine autour de la déchirure rétinienne au bord du colobome pour renforcer l’adhésion ; l’acuité visuelle finale s’est améliorée à 20/50.
Ratra et al. (2023) ont rapporté un cas de colobome choroïdien atypique compliqué d’une fistule sclérale post-traumatique, traité avec succès par vitrectomie, photocoagulation endoculaire et tamponnement gazeux 3).
Scemla et al. (2021) ont rapporté le cas d’un homme de 19 ans présentant une filtration transsclérale au niveau d’un colobome choroïdien, entraînant une hypotonie (4 mmHg) 1). La microscopie ultrasonique a confirmé un défaut scléral. La récupération spontanée est survenue en 6 semaines, avec une pression intraoculaire de 11 mmHg et une acuité visuelle de 1,0.