
Colobome de la papille optique
Points clés en un coup d’œil
Section intitulée « Points clés en un coup d’œil »1. Qu’est-ce que le colobome de la papille optique ?
Section intitulée « 1. Qu’est-ce que le colobome de la papille optique ? »Le colobome de la papille optique est une anomalie congénitale caractérisée par un élargissement anormal de la papille optique et une dépression blanche bien délimitée. Il résulte d’une fermeture incomplète de la fente optique (fente embryonnaire), qui se ferme normalement à la 7e semaine de gestation. Les vaisseaux rétiniens ne proviennent pas d’un seul point, mais de divers endroits sur le bord ou à l’intérieur de la dépression.
Lorsque la fermeture incomplète de la fente optique est limitée à la partie postérieure (côté du nerf optique), un colobome du nerf optique se produit. Si la fermeture incomplète est plus étendue de l’avant vers l’arrière, elle forme un spectre allant du colobome de l’iris à celui de la choroïde. Dans l’ensemble des colobomes, le colobome de la papille optique correspond à l’extrémité postérieure de la fermeture incomplète de la fente optique et fait partie du spectre continu avec le colobome de l’iris (extrémité antérieure), mais il existe également une forme localisée isolée de la papille optique.
Le code CIM-10 est H47.319 (nerf optique).
La distinction avec le syndrome de la fleur de liseron est importante. Dans le syndrome de la fleur de liseron, on observe un tissu de prolifération gliale au centre de la papille, avec des vaisseaux disposés radialement. Dans le colobome de la papille optique, il n’y a pas de prolifération gliale, la dépression est prédominante en bas et les vaisseaux proviennent de divers endroits sur le bord ou à l’intérieur de la dépression, ce qui permet la différenciation.
Le colobome de la papille optique présente une dépression blanche bien délimitée prédominante en bas, et les vaisseaux proviennent de divers endroits sur le bord ou à l’intérieur de la dépression. Il n’y a pas de tissu de prolifération gliale. Dans le syndrome de la fleur de liseron, il y a un tissu de prolifération gliale au centre de la papille, et les vaisseaux rayonnent à partir de la périphérie de la papille. Les deux sont des anomalies congénitales de la papille optique, mais ils peuvent être différenciés par l’examen du fond d’œil.
2. Principaux symptômes et signes cliniques
Section intitulée « 2. Principaux symptômes et signes cliniques »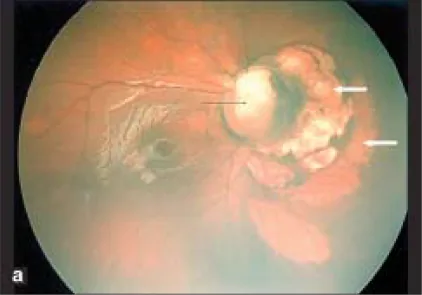
Symptômes subjectifs
Section intitulée « Symptômes subjectifs »L’acuité visuelle dépend de l’atteinte du faisceau papillo-maculaire par le colobome et de son degré. Elle varie de plus de 1,0 à des cas de mauvaise acuité, mais même en l’absence de lésion maculaire, l’acuité visuelle est souvent réduite en raison de l’anomalie du nerf optique.
- Baisse de l’acuité visuelle : dépend du degré d’atteinte du faisceau papillo-maculaire. Dans les cas de mauvaise acuité, un strabisme par non-usage peut survenir.
- Déficit du champ visuel : un déficit du champ visuel supérieur est fréquent, correspondant au colobome inférieur de la papille.
- Strabisme : dans les cas de mauvaise acuité visuelle, un strabisme par non-usage peut se développer.
Signes du fond d’œil
Section intitulée « Signes du fond d’œil »Au fond d’œil, on observe une perte de substance de la papille optique et de la choroïde et de la rétine inférieures, principalement en bas. Il existe des anomalies du trajet vasculaire ; l’artère centrale de la rétine se divise en arrière de la papille, de sorte que de nombreuses artères rétiniennes semblent émerger de la papille. Le bord supérieur de la papille est souvent conservé, et même lorsque l’ensemble est déprimé, la dépression est typiquement plus marquée en bas qu’en haut.
La région papillaire est déprimée, la papille est absente ou partiellement défectueuse, et la choroïde environnante, l’épithélium pigmentaire rétinien (EPR) et la sclère sont amincis. En dessous de la dépression papillaire, on observe une atrophie choroïdo-rétinienne et un aspect de fond d’œil tigré dus à une fermeture incomplète de la fente embryonnaire.
Classification
Section intitulée « Classification »Le colobome de la papille optique est classé comme suit en fonction de l’étendue de l’atteinte :
- Colobome papillaire isolé : dû à une fermeture incomplète localisée de la partie postérieure de la fente optique.
- Colobome choroïdo-rétinien associé : indique une fermeture incomplète plus étendue de la fente optique.
- Type combiné colobome irien et ciliaire : forme étendue continue de l’extrémité antérieure à l’extrémité postérieure.
- Colobome de Fuchs : forme légère, présentant une lésion atrophique similaire à un cône en dessous de la papille. La vision est souvent relativement préservée.
Complications
Section intitulée « Complications »Complications oculaires
Section intitulée « Complications oculaires »- Souvent associé à un colobome irien et un colobome choroïdien.
- En cas de colobome choroïdien associé avec une large zone de dépression, une microphtalmie peut survenir.
- Décollement séreux de la rétine : peut survenir même avec un colobome isolé de la papille optique.
- Décollement rhegmatogène de la rétine : peut survenir secondairement dans les cas de colobome choroïdien complexe.
- Hypotonie due à une filtration transsclérale : des cas de fuite d’humeur aqueuse à travers le défaut scléral ont été rapportés 7).
3. Causes, épidémiologie et facteurs de risque
Section intitulée « 3. Causes, épidémiologie et facteurs de risque »Épidémiologie
Section intitulée « Épidémiologie »La prévalence est rapportée entre 3 et 8/100 000. Les cas unilatéraux et bilatéraux sont également fréquents, avec une majorité de cas sporadiques, mais souvent des antécédents familiaux. Divers modes de transmission (autosomique dominant, autosomique récessif, lié à l’X) ont été rapportés 4).
Le taux de diagnostic génétique global pour les colobomes reste inférieur à 30 % 5). De nombreux cas sporadiques sans mutation identifiée suggèrent l’implication de facteurs environnementaux et de multiples gènes modificateurs.
Mécanisme de développement
Section intitulée « Mécanisme de développement »La fissure fœtale (fissure optique) se forme ventralement lors de la formation de la cupule optique à partir du neuroectoderme à la 4e semaine de gestation. Elle se complète à la 5e semaine et commence à se fermer à partir de la 6e semaine. La fermeture progresse de la région équatoriale vers l’avant (côté irien) et vers l’arrière (côté du nerf optique) et s’achève à la 7e semaine. Un défaut de fermeture postérieure localisé entraîne un colobome du nerf optique.
Gènes associés
Section intitulée « Gènes associés »| Gène | Maladie associée | Remarques |
|---|---|---|
| PAX2 | Syndrome de colobome rénal (renal coloboma syndrome) | Impliqué dans la détermination ventrale de l’œil et la fermeture de la fissure embryonnaire1) |
| CHD7 | Syndrome CHARGE | Chromosome 8 (8q12.2), maladie rare désignée |
| FZD5 | Colobome syndromique + microphtalmie | Récepteur de la voie de signalisation Wnt2) |
Complications systémiques
Section intitulée « Complications systémiques »Le colobome de la papille optique peut être associé aux syndromes systémiques suivants.
- Syndrome CHARGE : syndrome de malformations multiples prenant les initiales de colobome (C), cardiopathie (H), atrésie des choanes (A), retard de croissance (R), hypoplasie génitale (G) et anomalies de l’oreille externe (E). Le gène CHD7 est le gène responsable, et il est reconnu comme maladie rare désignée.
- Syndrome d’Aicardi : associé à une agénésie du corps calleux, une épilepsie et un retard du développement psychomoteur. Survient principalement chez les filles. Le colobome se présente souvent sous forme de multiples lacunes dans la choroïde et la rétine.
- Syndrome colobome rénal : dû à une mutation du gène PAX2. Associé à des anomalies de la formation des voies urinaires rénales. Un suivi à long terme de la fonction rénale est nécessaire. La mutation frameshift c.76delG de PAX2 a été identifiée dans des familles atteintes de glomérulosclérose segmentaire et focale (FSGS), et le spectre phénotypique est plus large qu’on ne le pensait auparavant 1).
Il peut être associé à des syndromes systémiques tels que le syndrome CHARGE (malformations multiples dues à une mutation du gène CHD7, maladie rare désignée), le syndrome d’Aicardi (agénésie du corps calleux, épilepsie, prédominance féminine) et le syndrome colobome rénal (mutation PAX2, anomalies de la formation des voies urinaires rénales). En cas de bilatéralité ou de signes systémiques, une consultation pédiatrique et un conseil génétique sont recommandés.
4. Diagnostic et méthodes d’examen
Section intitulée « 4. Diagnostic et méthodes d’examen »Méthode diagnostique
Section intitulée « Méthode diagnostique »Le diagnostic est possible par simple ophtalmoscopie. Les points clés du diagnostic sont une dépression blanche bien délimitée prédominant dans la partie inférieure de la papille et une anomalie caractéristique du trajet vasculaire (nombreux vaisseaux partant du bord ou de l’intérieur de la dépression). Pour un diagnostic de certitude, on utilise l’échographie, l’IRM, le scanner et la tomographie par cohérence optique (OCT).
Bien que les cas sporadiques soient fréquents, des antécédents familiaux peuvent exister, d’où la nécessité d’une anamnèse familiale minutieuse.
Une IRM/CT cérébrale est nécessaire pour rechercher des malformations intracrâniennes associées (comme l’agénésie du corps calleux). Une consultation pédiatrique est réalisée pour vérifier l’absence de complications systémiques telles que le syndrome CHARGE ou le syndrome d’Aicardi.
| Examen | Objectif |
|---|---|
| Examen du fond d’œil (dilatation pupillaire) | Évaluation de la morphologie papillaire, du trajet vasculaire et du décollement de la rétine |
| OCT (tomographie par cohérence optique) | Évaluation détaillée de la structure de la papille optique et de la macula |
| Échographie (mode B) | Recherche de décollement de la rétine en cas de mauvaise visibilité du fond d’œil |
| Examen du champ visuel | Évaluation du motif de déficit du champ visuel (déficit du champ visuel supérieur, etc.) |
| IRM cérébrale | Recherche de complications du système nerveux central telles qu’agénésie du corps calleux et encéphalocèle |
| Échographie rénale | Dépistage du syndrome de colobome rénal |
| Test génétique | Recherche de mutations de PAX2, CHD7, etc. (en cas de bilatéralité ou de syndrome) |
Diagnostic différentiel
Section intitulée « Diagnostic différentiel »| Maladies différentielles | Points clés de différenciation |
|---|---|
| Syndrome de morning glory | Prolifération gliale au centre de la papille, vaisseaux en rayon. Dans le colobome, dépression inférieure sans prolifération gliale |
| Staphylome péripapillaire | Bombement postérieur de la sclère autour de la papille. Le colobome est une perte de substance de la papille elle-même |
| PFV/PHPV (persistance de l’artère hyaloïde primitive) | Avec tractus vitréen et plis rétiniens. Aspect du fond d’œil différent du colobome |
| Mégalopapille | Diamètre papillaire augmenté mais morphologie quasi normale. Pas d’excavation ni d’anomalie vasculaire 5) |
| Hypoplasie du nerf optique | Papille petite (rapport DM/DD ≥ 3,2). Le colobome présente une papille élargie et excavée |
| Atrophie optique glaucomateuse | Élargissement progressif de l’excavation et augmentation de la pression intraoculaire. Le colobome est non progressif avec pression normale |
5. Traitement standard
Section intitulée « 5. Traitement standard »Le colobome de la papille optique est une anomalie structurelle congénitale et il n’existe pas de traitement curatif. Le traitement est principalement symptomatique, en fonction de la présence et du type de complications.
Surveillance
Section intitulée « Surveillance »Étant donné qu’il s’agit d’une anomalie congénitale non progressive, en l’absence de complications telles qu’un décollement séreux de la rétine, une observation régulière du fond d’œil est poursuivie. Chez l’enfant, un examen du fond d’œil sous dilatation tous les six mois à un an est recommandé.
Prise en charge du décollement séreux de la rétine
Section intitulée « Prise en charge du décollement séreux de la rétine »Il n’existe pas de consensus sur le traitement du décollement séreux de la rétine, et des cas de régression spontanée ont été rapportés. Une période d’observation de quelques mois peut être envisagée. Si aucune amélioration n’est observée après la période de surveillance, une intervention chirurgicale est envisagée.
On suppose que l’anomalie structurelle de la zone excavée permet au liquide vitréen de pénétrer dans l’espace sous-rétinien, et la possibilité d’un écoulement de liquide céphalo-rachidien par une communication entre l’excavation et l’espace sous-arachnoïdien a également été suggérée.
Prise en charge du décollement rhegmatogène de la rétine
Section intitulée « Prise en charge du décollement rhegmatogène de la rétine »Pour le décollement rhegmatogène de la rétine, une vitrectomie et une photocoagulation autour de l’excavation sont réalisées. Le pronostic visuel postopératoire n’est pas toujours favorable.
Une technique de réapplication rétinienne utilisant de la colle de fibrine a été rapportée pour le décollement de rétine associé au colobome, consistant à appliquer de la colle de fibrine autour de la déchirure rétinienne au bord du colobome pour renforcer l’adhésion 3). Une amélioration de l’acuité visuelle finale à 20/50 a été obtenue dans certains cas.
Traitement de l’amblyopie
Section intitulée « Traitement de l’amblyopie »En cas de mauvaise acuité visuelle, en particulier unilatérale, une correction réfractive et une occlusion (de l’œil sain) sont réalisées. Une intervention précoce dans l’enfance est importante. Cependant, en cas de baisse de vision due à une anomalie structurelle du nerf optique lui-même, l’efficacité du traitement de l’amblyopie est limitée.
Prise en charge systémique
Section intitulée « Prise en charge systémique »- Cas associés au syndrome CHARGE : nécessité d’une prise en charge multidisciplinaire incluant chirurgie cardiaque, ORL et endocrinologie.
- Syndrome de colobome rénal : suivi à long terme de la fonction rénale.
- Conseil génétique : recommandé en cas de bilatéralité ou de syndrome.
Le décollement séreux de la rétine peut régresser spontanément, donc une observation de quelques mois est d’abord réalisée. Pour le décollement rhegmatogène, une vitrectomie et une photocoagulation autour de la dépression sont effectuées. Récemment, un renforcement de l’adhésion avec de la colle de fibrine a été rapporté. Cependant, le pronostic visuel postopératoire n’est pas toujours bon.
6. Physiopathologie et mécanismes détaillés
Section intitulée « 6. Physiopathologie et mécanismes détaillés »Processus de fermeture de la fissure fœtale
Section intitulée « Processus de fermeture de la fissure fœtale »La cupule optique se forme à partir du neuroectoderme à la 4e semaine de gestation. Une fissure fœtale (fissure optique) apparaît sur la face ventrale de la cupule optique, permettant le passage de l’artère hyaloïdienne. Cette fissure se complète à la 5e semaine et commence à se fermer à partir de la 6e semaine. La fermeture débute près de l’équateur et progresse vers l’avant (côté irien) et vers l’arrière (côté du nerf optique), se terminant à la 7e semaine. Un défaut de fermeture postérieure localisé entraîne un colobome du nerf optique.
La transition épithélio-mésenchymateuse (TEM) est impliquée dans le processus de fermeture. Les cellules épithéliales de la rétine neurale au bord de la fissure fœtale dégradent la membrane basale, acquièrent un phénotype mésenchymateux et fusionnent. Une perturbation de ce processus provoque un colobome6).
Mécanismes moléculaires
Section intitulée « Mécanismes moléculaires »Le gène PAX2 est impliqué dans la détermination ventrale de l’œil et dans la fermeture de la fissure fœtale. Les mutations de PAX2 provoquent le syndrome de colobome rénal. Une mutation par décalage du cadre de lecture c.76delG de PAX2 a été identifiée dans une famille atteinte de FSGS, et le phénotype du syndrome de colobome rénal est plus large qu’on ne le pensait auparavant1).
Le gène FZD5 code un récepteur de la voie de signalisation Wnt. Les mutations hypofonctionnelles altèrent l’activation dépendante du ligand de la signalisation Wnt, entraînant un défaut de fermeture de la fissure fœtale et une microcornée. Le mode de transmission est récessif2).
Le gène CHD7 code un facteur de remodelage de la chromatine et est impliqué dans la différenciation et la migration des cellules de la crête neurale. Les mutations provoquent le syndrome CHARGE.
Mécanisme du décollement séreux de la rétine
Section intitulée « Mécanisme du décollement séreux de la rétine »On suppose que le liquide vitréen s’écoule dans l’espace sous-rétinien en raison d’une anomalie structurelle de la zone excavée. La possibilité d’un écoulement de liquide céphalorachidien par une communication entre l’excavation et l’espace sous-arachnoïdien a également été suggérée, ce qui contribue à la difficulté de traiter le décollement séreux de la rétine.
Pronostic et évolution
Section intitulée « Pronostic et évolution »Le colobome de la papille optique est une anomalie congénitale fixe et non progressive. La complication d’un décollement de la rétine est le principal facteur aggravant le pronostic visuel. Même après une vitrectomie pour un décollement rhegmatogène de la rétine, le pronostic visuel est souvent peu favorable.
7. Recherches récentes et perspectives futures
Section intitulée « 7. Recherches récentes et perspectives futures »Élargissement du spectre phénotypique des mutations PAX2
Section intitulée « Élargissement du spectre phénotypique des mutations PAX2 »Hu et al. (2024) ont rapporté l’identification d’une mutation de décalage du cadre de lecture c.76delG dans PAX2 dans une famille atteinte de glomérulosclérose segmentaire et focale (FSGS) 1). Le phénotype du syndrome de colobome rénal est plus large qu’on ne le pensait auparavant, et l’importance du dépistage de la fonction rénale chez les patients présentant un colobome de la papille optique a été réaffirmée. Cela suggère d’élargir les indications de l’évaluation de la fonction rénale chez les patients présentant un colobome.
FZD5 et anomalies de la signalisation Wnt
Section intitulée « FZD5 et anomalies de la signalisation Wnt »Cortes-Gonzalez et al. (2024) ont rapporté qu’une mutation faux-sens homozygote de FZD5 (p.M160V) provoque un colobome oculaire symptomatique et une microcornée 2). L’analyse fonctionnelle a confirmé que l’activation de la signalisation Wnt dépendante du ligand est altérée selon un mode de transmission récessif. Le taux de diagnostic génétique du colobome est inférieur à 30 %, et l’identification de nouveaux gènes responsables devrait contribuer à améliorer le diagnostic.
Nouvelles techniques chirurgicales pour le décollement de la rétine
Section intitulée « Nouvelles techniques chirurgicales pour le décollement de la rétine »Jain et al. (2024) ont rapporté une amélioration de l’acuité visuelle finale à 20/50 après une rétinopexie avec colle de fibrine pour un décollement de la rétine associé à un colobome 3). La technique consistant à appliquer de la colle de fibrine autour de la déchirure rétinienne au bord du colobome pour renforcer l’adhésion est considérée comme une option complémentaire à la vitrectomie conventionnelle avec photocoagulation. L’accumulation de cas et la vérification des résultats à long terme sont des défis futurs.
8. Références
Section intitulée « 8. Références »-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.