La persistance du vitré primitif (persistent fetal vasculature: PFV) est une maladie oculaire congénitale due à une régression incomplète du système vasculaire vitréen fœtal. Anciennement appelée persistance de l’hyperplasie du vitré primitif (persistent hyperplastic primary vitreous: PHPV). En 1997, Goldberg a proposé le terme PFV pour inclure également les résidus de tissu fibrovasculaire autour du cristallin12), et ce terme est maintenant largement accepté.
Le système vasculaire vitréen comprend l’artère hyaloïde (hyaloid artery) issue de la papille optique et la tunique vasculaire du cristallin (tunica vasculosa lentis) en avant. Il se forme entre la 5e et la 6e semaine de gestation lorsque les cellules mésenchymateuses pénètrent dans la cavité vitréenne par la fente fœtale, atteignant son apogée à la 10e semaine. Ensuite, la régression commence à partir de la périphérie entre la 13e et la 15e semaine et se termine à la fin de la période fœtale. Dans la PFV, cette régression est incomplète, entraînant des anomalies du développement des tissus périvasculaires.
Elle est considérée comme unilatérale et non héréditaire, et aucun gène spécifique n’a été identifié comme cause. Les cas typiques se présentent avec une leucocorie associée à une microphtalmie, mais peuvent également être diagnostiqués à l’occasion d’un strabisme ou d’une amblyopie.
La PFV est associée à environ 20 % des cataractes du nourrisson et de l’enfant 2)3). Dans le registre des cataractes du Pediatric Eye Disease Investigator Group (PEDIG), 64 des 994 cas (6,4 %) étaient des cataractes associées à une PFV 1), dont 75 % ont été opérés comme aphakes (âge médian à la chirurgie : 2 mois) et 25 % comme pseudophakes (âge médian : 29 mois) 1). Des anomalies du segment postérieur (anomalies du vitré, de la rétine ou du nerf optique) ont été observées dans 28 % des cas 1), et seulement 4 à 6 % avaient des antécédents familiaux 1).
QQuelle est la différence entre PFV et PHPV ?
A
Il s’agit essentiellement d’une différence de terminologie entre l’ancien et le nouveau nom pour la même maladie. PHPV est l’ancienne appellation, mettant l’accent sur l’hyperplasie et la persistance du système hyaloïde (vitré primitif). En 1997, Goldberg a proposé le terme PFV (persistent fetal vasculature) pour inclure également la persistance de tissu fibrovasculaire autour du cristallin. Le PFV permet de décrire un spectre lésionnel plus large et est désormais le terme standard.
QLe PFV survient-il dans les deux yeux ?
A
Le PFV est presque toujours unilatéral et considéré comme non héréditaire. Dans l’étude PEDIG, seuls 3 cas de PFV bilatéral ont été exclus 1). En cas de bilatéralité, un diagnostic différentiel avec la vitréorétinopathie exsudative familiale (FEVR) ou la maladie de Norrie est nécessaire, et un bilan incluant des tests génétiques est recommandé.
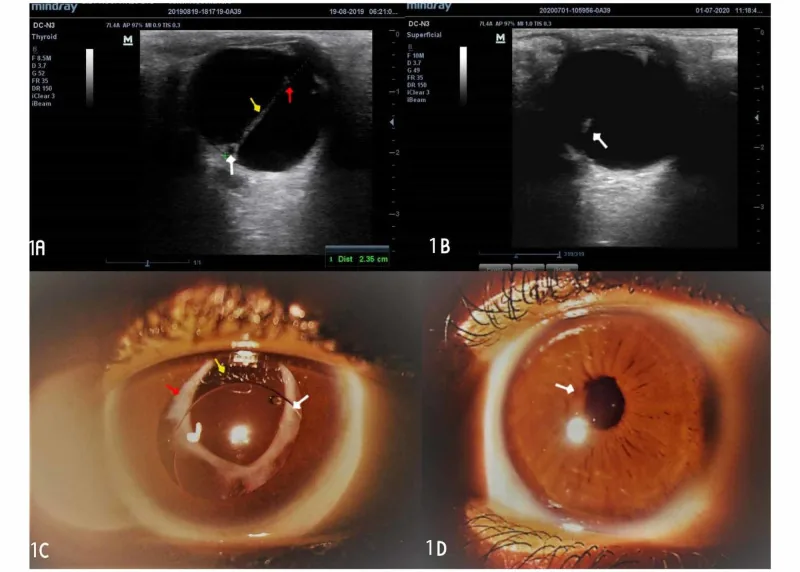
Egbu E The Outcome of Manual Small Incision Cataract Surgery and Anterior Vitrectomy for Persistent Fetal Vasculature in an 18-Year-Old Woman: A One-Year Follow-Up. Cureus. 2020. Figure 1. PMCID: PMC7584328. License: CC BY.
L’échographie oculaire montre une bande hyperéchogène traversant le vitré, et la photographie du segment antérieur révèle, sous mydriase, un tissu fibrovasculaire blanc en arrière de la pupille. Ces images illustrent la membrane fibrovasculaire rétrolentale et la bande vitréenne caractéristiques du PFV, appropriées pour la section des principaux symptômes et signes cliniques.
Le PFV est classé en trois types selon la localisation prédominante des lésions : antérieur, postérieur et mixte.
Type antérieur
Principaux signes : Membrane fibroproliférative sur la face postérieure du cristallin (tache de Mittendorf), cataracte sous-capsulaire postérieure, lenticône postérieur
Indication thérapeutique : Chirurgie indiquée si la lésion est limitée à la partie postérieure du cristallin et que le fond d’œil est normal
Pronostic : Le meilleur des trois types. Acuité visuelle médiane 20/100 (acuité ≥20/200 dans 54% des cas) 1)
Type postérieur
Principaux signes : Bande allant de la papille vers le vitré, pli rétinien, décollement de rétine, dysplasie rétinienne
Indications thérapeutiques : Si l’ERG/VEP est bon, envisager une intervention chirurgicale. Aucun traitement efficace pour la dysplasie rétinienne.
Pronostic : Défavorable. Acuité visuelle médiane 20/800 (acuité visuelle ≥20/200 dans 36% des cas)1)
Type mixte
Principales observations : Mélange de signes de type antérieur et postérieur. Souvent associé à une microphtalmie.
Indications thérapeutiques : Décision prudente en fonction de l’étendue des lésions individuelles.
Pronostic : Dans les formes mixtes sévères, seulement 19% ont une acuité visuelle mesurable, 61% n’ont pas de perception lumineuse8)
Classification détaillée des 7 types en ophtalmologie pédiatrique
Les principales constatations comprennent une microphtalmie unilatérale, une opacité de la face postérieure du cristallin (tache de Mittendorf), une anomalie du trajet des vaisseaux rétiniens, une dysplasie rétinienne, des procès ciliaires allongés et un cordon s’étendant de la face postérieure du cristallin à la papille optique. Le cordon peut provoquer une traction et, via l’allongement des procès ciliaires, entraîner une hypotonie. La rétine peut être impliquée dans le tissu cordonal près de la papille. La sévérité varie selon le degré de régression de la vascularisation vitréenne.
Dans l’étude PEDIG, 28 % (18/64 yeux) des cataractes PFV présentaient des anomalies du segment postérieur 1), et 46 % des yeux pseudophakes avaient une pathologie du segment postérieur (OR ajusté sur l’âge 4,47 par rapport à 28 % des yeux aphakes) 1). Une vitrectomie antérieure a été réalisée lors de la chirurgie de la cataracte dans 96 % des yeux PFV aphakes 1).
Au cours du développement normal du système vasculaire vitréen, les cellules mésenchymateuses pénètrent dans la cavité vitréenne par la fissure embryonnaire à la 5e-6e semaine de gestation, formant le système vasculaire vitréen. Il atteint son apogée à la 10e semaine, puis la régression commence à partir de la périphérie. Plus précisément, la régression débute à la 13e-15e semaine et se termine à la fin de la période fœtale.
Dans la PFV, une régression incomplète de la vascularisation vitréenne se produit, entraînant des anomalies du développement des tissus périvasculaires. Cela conduit à une opacité cristallinienne, une dysplasie rétinienne et une microphtalmie. De plus, la traction du tissu résiduel provoque un allongement des procès ciliaires et des plis rétiniens.
La PFV est considérée comme unilatérale et non héréditaire, et aucun gène spécifique n’a été identifié comme cause. Seuls 4 à 6 % des cas ont des antécédents familiaux 1), la plupart étant sporadiques. En cas de bilatéralité, un diagnostic génétique différentiel avec la FEVR (mutations des gènes NDP, LRP5, FZD4) ou la maladie de Norrie (mutation du gène NDP) est important.
Aucune association claire n’a été démontrée avec un faible poids de naissance ou une prématurité, mais une persistance sévère de la vascularisation vitréenne peut rarement présenter des similitudes avec la rétinopathie du prématuré.
Échographie (B-scan) : visualise les cordons rétrolentaux. Utile pour évaluer la microphtalmie et la cavité vitréenne. Particulièrement importante lorsque les milieux transparents sont opaques, car la région postérieure n’est pas directement visible.
TDM : détecte la présence de calcifications intraoculaires. Le rétinoblastome présente des calcifications, contrairement à la PFV, ce qui en fait l’examen le plus utile pour différencier les deux maladies.
IRM : excellente pour évaluer les cordons, les anomalies du segment postérieur et l’infiltration du nerf optique.
ERG et VEP (électrorétinogramme et potentiels évoqués visuels) : évaluation de la fonction rétinienne. Permet d’évaluer la fonction visuelle avant l’opération et de déterminer l’indication d’une intervention chirurgicale.
La distinction avec les maladies provoquant une leucocorie est primordiale. En particulier, le diagnostic différentiel avec le rétinoblastome a un impact direct sur le pronostic vital.
Diagnostic différentiel avec le rétinoblastome
Maladie à exclure en priorité absolue
La PFV s’accompagne généralement d’une microphtalmie, contrairement au rétinoblastome. La TDM est utile pour détecter les calcifications intraoculaires, caractéristiques du rétinoblastome et absentes dans la PFV.
Diagnostic différentiel avec la FEVR et la maladie de Norrie
Important en cas d’atteinte bilatérale
En cas de décollement total de la rétine ou de prolifération fibreuse rétrolentale bilatérale, il faut envisager un diagnostic différentiel avec la FEVR ou la maladie de Norrie. Les antécédents familiaux et la recherche d’anomalies génétiques sont utiles. L’utilisation de panels de séquençage de nouvelle génération (NGS) est utile.
Zone rétinienne périphérique avasculaire, prolifération vasculaire
QQuelle est la maladie à exclure en priorité en cas de leucocorie ?
A
Le rétinoblastome. C’est la tumeur maligne intraoculaire la plus fréquente chez l’enfant et elle a un impact direct sur le pronostic vital, d’où la nécessité d’une exclusion rapide. La PFV s’accompagne souvent d’une microphtalmie, alors que le rétinoblastome ne présente généralement pas de microphtalmie. La recherche de calcifications intraoculaires par tomodensitométrie est l’étape de diagnostic différentiel la plus importante. Si des calcifications sont confirmées, un rétinoblastome est fortement suspecté et une orientation vers un centre spécialisé est nécessaire.
L’opacité du cristallin est traitée de la même manière que la cataracte. Si elle est localisée à la partie postérieure du cristallin et que le fond d’œil est normal, on procède à une ablation du cristallin et à une excision de la membrane fibroproliférative, comme pour la cataracte congénitale. Si les bandes à l’arrière du cristallin sont excentrées et n’affectent pas l’axe visuel, la chirurgie n’est pas nécessaire. Si la lésion s’étend au fond d’œil, la chirurgie n’est généralement pas indiquée. En cas de décollement de la rétine ou de traction, une bonne réponse à l’ERG/VEP justifie une intervention chirurgicale. En cas de déformation ou de fermeture pupillaire due à une prolifération antérieure du cristallin, une pupilloplastie est réalisée.
Après l’ablation du cristallin, on procède à la correction de la réfraction et au traitement de l’amblyopie. La technique chirurgicale est la même que pour la cataracte congénitale. Dans l’étude PEDIG, 96 % des yeux PFV aphakes ont subi une vitrectomie antérieure lors de la chirurgie de la cataracte1). L’incidence de l’opacification de l’axe visuel est de 18 % lorsque la vitrectomie antérieure est réalisée lors de la première intervention, contre 60 % lorsqu’elle ne l’est pas 1).
Après la chirurgie, un traitement de l’amblyopie est effectué, combinant correction de la réfraction (lentilles de contact ou lunettes) et occlusion de l’œil sain (patch). Dans le PFV unilatéral, l’observance du traitement de l’amblyopie influence le pronostic visuel. Le traitement doit être actif pendant la période critique du développement visuel (de la naissance à environ 10 ans).
Résultats chirurgicaux (résultats à 5 ans de l’étude PEDIG sur la cataracte PFV)
Événements indésirables liés au glaucome (cumul à 5 ans)
24 % (IC à 95 % : 9–37 %)
7 % (IC à 95 % : 0–20 %)
Opacification de l’axe visuel
15 % (IC à 95 % : 5–25 %)
45 % (IC à 95 % : 13–66 %)
Décollement de la rétine
4 % (IC à 95 % : 0–10 %)
7 % (IC à 95 % : 0–19 %)
En ce qui concerne les résultats visuels globaux, 4 yeux sur 42 (10 %, IC à 95 % 3–23 %) ont atteint une acuité visuelle normale pour l’âge, et 48 % (IC à 95 % 32–64 %) ont atteint une acuité visuelle de 20/200 ou mieux 1). Le taux d’atteinte de 20/200 ou mieux dans les yeux PFV pseudophakes (23 %) était significativement inférieur à celui des yeux pseudophakes non PFV (68 %) (OR ajusté pour l’âge = 0,14, P = 0,005) 1). Les yeux PFV aphakes ont montré des résultats similaires par rapport aux yeux aphakes non PFV (OR ajusté pour l’âge = 1,90, P = 0,14) 1).
Selon le type, l’acuité visuelle médiane dans le type antérieur était de 20/100 (54 % avec une acuité de 20/200 ou mieux), tandis que dans le type postérieur, l’acuité visuelle médiane était plus faible, à 20/800 (36 % avec une acuité de 20/200 ou mieux), avec une différence de 4 lignes logMAR (P = 0,09) 1).
Dans d’autres rapports, l’étude monocentrique de 20 ans de Bata et al. (58 yeux PFV aphakes) a montré que 33 % avaient une acuité visuelle de 20/200 ou mieux (suivi moyen de 6,7 ans) 4), le rapport d’Anteby et al. (30 yeux PFV aphakes) a montré que 16,7 % avaient une acuité de 20/200 ou mieux (suivi moyen de 8,5 ans) 5), et de Saint Sauveur et al. ont rapporté que sur 36 cas de PFV mixte sévère, seulement 19 % avaient une acuité visuelle mesurable et 61 % n’avaient pas de perception lumineuse 8).
Les autres complications majeures incluent l’opacification de l’axe visuel (45 % des yeux PFV pseudophakes, 15 % des yeux aphakes), et le taux cumulé de chirurgie de clarification de l’axe visuel atteint 40 % dans les yeux PFV pseudophakes 1). De plus, 13 % (IC à 95 % 2–22 %) des yeux PFV aphakes nécessitent une implantation secondaire de LIO dans les 5 ans 1).
Prise en charge des cas graves et du type postérieur
Il n’existe pas de traitement efficace pour la dysplasie rétinienne. Dans le type postérieur, le développement visuel est souvent peu probable. En cas de microphtalmie marquée, le port précoce d’une prothèse oculaire est envisagé pour des raisons esthétiques. Le port d’une prothèse oculaire contribue également à la croissance orbitaire.
QQuelle acuité visuelle peut-on attendre après une chirurgie PFV ?
A
Dans le type antérieur (limité à la partie postérieure du cristallin), l’acuité visuelle médiane à 5 ans postopératoires est de 20/100, avec un taux d’atteinte de 20/200 ou mieux de 54 % 1). Cependant, seulement 10 % de l’ensemble des patients atteignent une acuité visuelle normale pour l’âge. Dans le type postérieur, l’acuité visuelle médiane est faible, à 20/800 1). Le pronostic visuel est fortement influencé par le type de PFV, la présence de lésions du segment postérieur, l’âge chirurgical et l’observance du traitement de l’amblyopie.
QQuelle est la complication la plus importante à surveiller après une chirurgie PFV ?
A
Les événements indésirables liés au glaucome sont les plus fréquents, atteignant un taux cumulatif de 24 % à 5 ans dans les yeux PFV aphakes 1). L’opacité de l’axe visuel est également une complication importante, survenant dans 45 % des yeux pseudophakes 1). La vitrectomie antérieure lors de la première chirurgie est importante pour prévenir l’opacité de l’axe visuel (18 % avec vs 60 % sans) 1). Après la chirurgie, une gestion à long terme de la pression intraoculaire, une évaluation de l’axe visuel et une correction réfractive sont nécessaires.
À la 5e-6e semaine de gestation, les cellules mésenchymateuses pénètrent dans la cavité vitréenne par la fissure optique, formant le système vasculaire vitréen composé de l’artère hyaloïde et de la tunique vasculaire du cristallin. Ce système vasculaire joue un rôle crucial en fournissant oxygène et nutriments au cristallin en développement et au segment antérieur de l’œil.
La 10e semaine de gestation est le pic de développement, après quoi la régression commence à partir de la périphérie. La régression s’intensifie de la 13e à la 15e semaine et est presque complète à la fin de la gestation. À la naissance normale, le système vasculaire vitréen est presque absent, bien qu’une trace puisse subsister sous forme de tache de Mittendorf (petit point blanc sur la face postérieure du cristallin).
Dans le PFV, il y a une régression incomplète des vaisseaux vitréens, entraînant une anomalie de développement du tissu fibroprolifératif périvasculaire. Le tissu mésenchymateux résiduel autour des vaisseaux prolifère et se fibrose, donnant lieu à diverses manifestations cliniques.
Les conséquences spécifiques sont les suivantes :
Opacité cristallinienne (cataracte sous-capsulaire postérieure, lenticône postérieur) : adhérence du tissu fibreux résiduel à la face postérieure du cristallin
Microphtalmie : trouble du développement global de l’œil dû à une anomalie du développement du tissu périvasculaire
Dysplasie rétinienne : dans la forme postérieure, dysplasie rétinienne due à la traction et à l’ischémie
Allongement des procès ciliaires et hypotonie : traction par les cordons fibreux
Plis rétiniens : déformation due à la traction du tissu pathologique postérieur
La raison pour laquelle elle est unilatérale et non héréditaire est que la régression des vaisseaux vitréens fœtaux est contrôlée par des signaux locaux d’angiogenèse et de régression, et on suppose qu’une perturbation de ce mécanisme de contrôle ne survient que dans un seul œil. Cependant, le mécanisme moléculaire précis n’est pas encore élucidé.
Le gène spécifique responsable de la PFV n’a pas encore été identifié, mais des modèles animaux ont montré que des mutations dans les gènes liés à la voie de signalisation Wnt, tels que FZD4, LRP5 et NDP, présentent un phénotype de type PFV (persistance du vitré primitif). Ces gènes sont également responsables de la FEVR et de la maladie de Norrie, suggérant une continuité génétique entre la PFV et les maladies apparentées. L’application clinique des tests par panel de séquençage de nouvelle génération (NGS) pour le diagnostic différentiel de la PFV progresse également.
Des approches de la PFV postérieure par vitrectomie micro-incisionnelle (MIVS) de calibre 25 ou 27 sont tentées. La chirurgie mini-invasive chez les nouveau-nés et les nourrissons présente l’avantage de minimiser le traumatisme et de réduire l’inflammation postopératoire11).
La comparaison des résultats à long terme entre l’implantation primaire de la LIO pendant la petite enfance et l’implantation secondaire après gestion de l’aphakie est une question importante dans la recherche sur la PFV. Les données de suivi prolongé de l’étude PEDIG devraient permettre d’établir des critères d’indication spécifiques à la PFV pour l’implantation de la LIO6)7).
L’évaluation non invasive de la PFV par caméra grand angle du fond d’œil et OCT du segment antérieur progresse. L’OCT du segment antérieur est particulièrement utile pour l’évaluation morphologique du lenticône postérieur et de la cataracte sous-capsulaire postérieure, ainsi que pour la planification préopératoire.
Haider KM, Repka MX, Sutherland DR, Hatt SR, Fallaha N, Kraker RT, et al. Outcomes and Complications 5 Years After Surgery for Pediatric Cataract Associated With Persistent Fetal Vasculature. American journal of ophthalmology. 2024;260:30-36. doi:10.1016/j.ajo.2023.11.002. PMID:37939986; PMCID:PMC11005992.
Wilson ME, Trivedi RH, Morrison DG, Lambert SR, Buckley EG, Plager DA, et al. The Infant Aphakia Treatment Study: evaluation of cataract morphology in eyes with monocular cataracts. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2011;15(5):421-6. doi:10.1016/j.jaapos.2011.05.016. PMID:22108352; PMCID:PMC3345197.
Solebo AL, Russell-Eggitt I, Cumberland P, Rahi JS. Congenital cataract associated with persistent fetal vasculature: findings from IoLunder2. Eye (London, England). 2016;30(9):1204-9. doi:10.1038/eye.2016.159. PMID:27472205; PMCID:PMC5023809.
Bata BM, Chiu HH, Mireskandari K, Ali A, Lam WC, Wan MJ. Long-term visual and anatomic outcomes following early surgery for persistent fetal vasculature: a single-center, 20-year review. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2019;23(6):327.e1-327.e5. doi:10.1016/j.jaapos.2019.07.009. PMID:31629823.
Anteby I, Cohen E, Karshai I, BenEzra D. Unilateral persistent hyperplastic primary vitreous: course and outcome. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2002;6(2):92-9. doi:10.1067/mpa.2002.121324. PMID:11997805.
Repka MX, Dean TW, Lazar EL, Yen KG, Lenhart PD, Freedman SF, et al. Cataract Surgery in Children from Birth to Less than 13 Years of Age: Baseline Characteristics of the Cohort. Ophthalmology. 2016;123(12):2462-2473. doi:10.1016/j.ophtha.2016.09.003. PMID:27769584; PMCID:PMC5121052.
Repka MX, Dean TW, Kraker RT, Li Z, Yen KG, de Alba Campomanes AG, Young MP, Rahmani B, et al. Visual Acuity and Ophthalmic Outcomes 5 Years After Cataract Surgery Among Children Younger Than 13 Years. JAMA ophthalmology. 2022;140(3):269-276. doi:10.1001/jamaophthalmol.2021.6176. PMID:35142808; PMCID:PMC8832311.
de Saint Sauveur G, Chapron T, Abdelmassih Y, Chehaibou I, Lecler A, Dureau P, et al. Management and Outcomes of Posterior Persistent Fetal Vasculature. Ophthalmology. 2023;130(8):844-853. doi:10.1016/j.ophtha.2023.03.027. PMID:37044159.
Khandwala N, Besirli C, Bohnsack BL.. Outcomes and surgical management of persistent fetal vasculature. BMJ Open Ophthalmol. 2021;6(1):e000656. doi:10.1136/bmjophth-2020-000656. PMID:34013048; PMCID:PMC8094357.
Hunt A, Rowe N, Lam A, Martin F.. Outcomes in persistent hyperplastic primary vitreous. Br J Ophthalmol. 2005;89(7):859-863. doi:10.1136/bjo.2004.053595. PMID:15965167; PMCID:PMC1772745.
Bata BM, Khalili S, Ali A, Wan MJ, Mireskandari K. Late surgery for unilateral persistent fetal vasculature: long-term visual and anatomic outcomes. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2022;26(6):296.e1-296.e5. doi:10.1016/j.jaapos.2022.09.005. PMID:36265751.
Goldberg MF. Persistent fetal vasculature (PFV): an integrated interpretation of signs and symptoms associated with persistent hyperplastic primary vitreous (PHPV). LIV Edward Jackson Memorial Lecture. American journal of ophthalmology. 1997;124(5):587-626. doi:10.1016/s0002-9394(14)70899-2. PMID:9372715.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.