La persistenza del vitreo primitivo (persistent fetal vasculature: PFV) è una malattia oculare congenita causata da un’incompleta regressione del sistema vascolare vitreale fetale. In precedenza era chiamata persistenza dell’iperplasia del vitreo primario (persistent hyperplastic primary vitreous: PHPV). Nel 1997, Goldberg propose il termine PFV per includere anche i residui di tessuto fibrovascolare intorno al cristallino12), e ora è ampiamente accettato.
Il sistema vascolare vitreale è costituito dall’arteria ialoidea (hyaloid artery) che origina dalla papilla ottica e dalla tunica vascolosa del cristallino (tunica vasculosa lentis) anteriormente. Si forma tra la 5a e la 6a settimana di gestazione quando le cellule mesenchimali entrano nella cavità vitreale attraverso la fessura fetale, raggiungendo il picco alla 10a settimana. Successivamente, la regressione inizia dalla periferia tra la 13a e la 15a settimana e termina nel periodo fetale tardivo. Nella PFV, questa regressione è incompleta, causando anomalie dello sviluppo dei tessuti perivascolari.
È considerata unilaterale e non ereditaria, e non è stato identificato alcun gene specifico come causa. I casi tipici si presentano con leucocoria associata a microftalmia, ma possono anche essere diagnosticati in occasione di strabismo o ambliopia.
La PFV è associata a circa il 20% delle cataratte infantili e pediatriche 2)3). Nel registro delle cataratte del Pediatric Eye Disease Investigator Group (PEDIG), 64 dei 994 casi (6,4%) erano cataratte associate a PFV1), di cui il 75% è stato operato come afachico (età mediana all’intervento 2 mesi) e il 25% come pseudofachico (età mediana 29 mesi) 1). Il 28% presentava anomalie del segmento posteriore (anomalie del vitreo, della retina o del nervo ottico) 1), e solo il 4-6% aveva una storia familiare 1).
QQual è la differenza tra PFV e PHPV?
A
Si tratta essenzialmente di una differenza tra il vecchio e il nuovo nome della stessa malattia. PHPV è la vecchia denominazione, che si concentrava sull’iperplasia e la persistenza dei vasi ialoidei (vitreo primario). Nel 1997, Goldberg propose il termine PFV (persistent fetal vasculature) per includere anche la persistenza di tessuto fibrovascolare intorno al cristallino. Il PFV può descrivere uno spettro lesionale più ampio ed è ora il termine standard.
QIl PFV si verifica in entrambi gli occhi?
A
Il PFV è quasi sempre unilaterale e considerato non ereditario. Nello studio PEDIG, solo 3 casi di PFV bilaterale sono stati esclusi 1). In caso di bilateralità, è necessaria una diagnosi differenziale con la vitreoretinopatia essudativa familiare (FEVR) e la malattia di Norrie, e si raccomanda un approfondimento che includa test genetici.
Egbu E The Outcome of Manual Small Incision Cataract Surgery and Anterior Vitrectomy for Persistent Fetal Vasculature in an 18-Year-Old Woman: A One-Year Follow-Up. Cureus. 2020. Figure 1. PMCID: PMC7584328. License: CC BY.
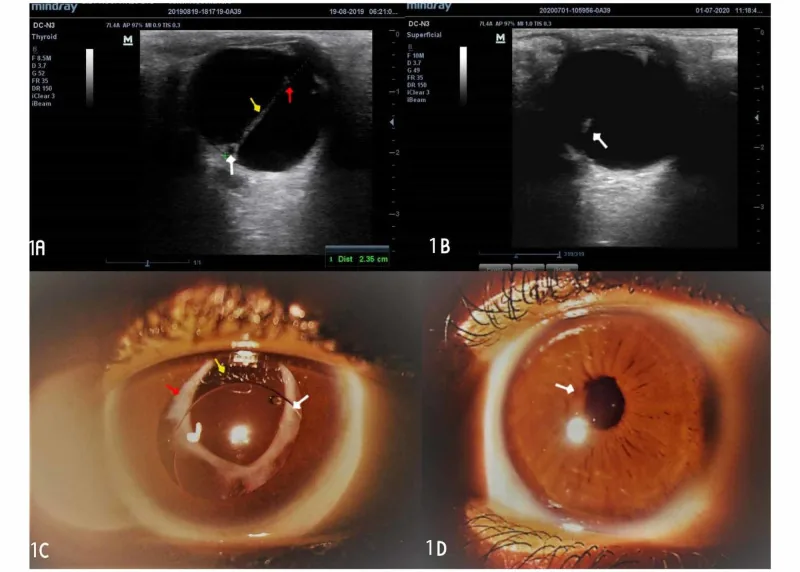
L’ecografia oculare mostra una lesione cordonale iperecogena che attraversa il vitreo, e la fotografia del segmento anteriore in midriasi rivela tessuto fibrovascolare bianco dietro la pupilla. Questi reperti, che illustrano la membrana fibrovascolare retrolenticolare e le bande vitreali caratteristiche del PFV, sono adatti per la sezione dei principali sintomi e segni clinici.
Il PFV è classificato in tre tipi in base alla localizzazione predominante delle lesioni: tipo anteriore, tipo posteriore e tipo misto.
Tipo anteriore
Principali reperti : Membrana fibroproliferativa sulla superficie posteriore del cristallino (macchia di Mittendorf), cataratta sottocapsulare posteriore, lenticono posteriore
Indicazione terapeutica : Chirurgia indicata se la lesione è limitata alla parte posteriore del cristallino e il fondo è normale
Prognosi : La migliore dei tre tipi. Acuità visiva mediana 20/100 (acuità ≥20/200 nel 54% dei casi) 1)
Tipo posteriore
Principali reperti : Banda dalla papilla al vitreo, piega retinica, distacco di retina, displasia retinica
Indicazioni terapeutiche: Se ERG/VEP sono buoni, considerare l’intervento chirurgico. Nessun trattamento efficace per la displasia retinica.
I principali reperti includono microftalmia unilaterale, opacità della superficie posteriore del cristallino (macchia di Mittendorf), decorso anomalo dei vasi retinici, displasia retinica, processi ciliari allungati e un cordone che si estende dalla superficie posteriore del cristallino alla papilla ottica. Il cordone può causare trazione e, attraverso l’allungamento dei processi ciliari, portare a ipotonia. La retina può essere coinvolta nel tessuto cordonale vicino alla papilla. La gravità varia in base al grado di regressione dei vasi vitreali.
Nello studio PEDIG, il 28% (18/64 occhi) delle cataratte PFV presentava anomalie del segmento posteriore 1) e il 46% degli occhi pseudofachici presentava patologia del segmento posteriore (OR aggiustato per età 4,47 rispetto al 28% degli occhi afachici) 1). Nel 96% degli occhi PFV afachici è stata eseguita una vitrectomia anteriore al momento dell’intervento di cataratta1).
Nello sviluppo normale del sistema vascolare vitreale, alla 5a-6a settimana di gestazione le cellule mesenchimali entrano nella cavità vitrea attraverso la fessura embrionale e formano il sistema vascolare vitreale. Raggiunge il suo apice alla 10a settimana, dopodiché inizia la regressione dalla periferia. Nello specifico, la regressione inizia alla 13a-15a settimana e si attenua nel periodo fetale tardivo.
Nella PFV si verifica una regressione incompleta dei vasi vitreali, che porta ad anomalie dello sviluppo del tessuto perivascolare. Ciò provoca opacità del cristallino, displasia retinica e microftalmia. Inoltre, la trazione del tessuto residuo causa allungamento dei processi ciliari e pieghe retiniche.
La PFV è considerata unilaterale e non ereditaria, e non è stato identificato alcun gene specifico come causa. Solo il 4-6% ha una storia familiare 1), la maggior parte sono sporadici. In caso di bilateralità, è importante la diagnosi differenziale genetica con FEVR (mutazioni dei geni NDP, LRP5, FZD4) e malattia di Norrie (mutazione del gene NDP).
Non è stata dimostrata una chiara associazione con basso peso alla nascita o prematurità, ma gravi residui vascolari vitreali possono raramente presentare reperti simili alla retinopatia del prematuro.
Ecografia (B-scan): visualizza i cordoni retro-lenticolari. Utile per valutare la microftalmia e la cavità vitreale. Particolarmente importante quando i mezzi trasparenti sono opachi, poiché il polo posteriore non è direttamente visibile.
TC: rileva la presenza di calcificazioni intraoculari. Il retinoblastoma presenta calcificazioni, mentre la PFV no, pertanto è l’esame più utile per differenziare le due malattie.
RM: eccellente per valutare cordoni, anomalie del segmento posteriore e infiltrazione del nervo ottico.
ERG e VEP (elettroretinogramma e potenziali evocati visivi): valutazione della funzione retinica. Consente di valutare la funzione visiva prima dell’intervento e di determinare l’opportunità di un intervento chirurgico.
La differenziazione dalle malattie che causano leucocoria è di fondamentale importanza. In particolare, la diagnosi differenziale con il retinoblastoma ha un impatto diretto sulla prognosi vitale.
Diagnosi differenziale con il retinoblastoma
Malattia da escludere con la massima priorità
La PFV è solitamente accompagnata da microftalmia, mentre il retinoblastoma no. La TC per rilevare calcificazioni intraoculari è utile; la calcificazione è caratteristica del retinoblastoma e assente nella PFV.
Diagnosi differenziale con FEVR e malattia di Norrie
Importante in caso di bilateralità
In caso di distacco retinico totale bilaterale o proliferazione fibrosa retro-lenticolare, è necessaria la diagnosi differenziale con FEVR o malattia di Norrie. L’anamnesi familiare e la ricerca di anomalie genetiche sono utili. L’uso di pannelli di sequenziamento di nuova generazione (NGS) è utile.
Area retinica periferica avascolare, proliferazione vascolare
QQuale malattia deve essere esclusa per prima in caso di leucocoria?
A
Il retinoblastoma. È il tumore maligno intraoculare più comune nei bambini e ha un impatto diretto sulla prognosi di sopravvivenza, quindi è necessaria una rapida esclusione. La PFV è spesso associata a microftalmia, mentre nel retinoblastoma di solito non si osserva microftalmia. La TC per verificare la presenza di calcificazioni intraoculari è il passo differenziale più importante. Se le calcificazioni sono confermate, si sospetta fortemente un retinoblastoma ed è necessario un rinvio a un centro specialistico.
L’opacità del cristallino viene trattata come la cataratta. Se è localizzata nella parte posteriore del cristallino e il fondo è normale, si procede con lensectomia ed escissione della membrana fibroproliferativa, come per la cataratta congenita. Se le bande posteriori del cristallino sono eccentriche e non interessano l’asse visivo, l’intervento chirurgico non è necessario. Se la lesione si estende al fondo, generalmente la chirurgia non è indicata. In caso di distacco di retina o trazione, una buona risposta all’ERG/VEP costituisce la base per l’intervento chirurgico. In caso di deformazione o chiusura pupillare dovuta a proliferazione anteriore del cristallino, si esegue una pupilloplastica.
Dopo la rimozione del cristallino, si procede con la correzione refrattiva e il trattamento dell’ambliopia. La tecnica chirurgica è la stessa della cataratta congenita. Nello studio PEDIG, il 96% degli occhi afachici con PFV è stato sottoposto a vitrectomia anteriore al momento dell’intervento di cataratta1). L’incidenza di opacizzazione dell’asse visivo è stata del 18% quando la vitrectomia anteriore è stata eseguita al primo intervento, rispetto al 60% quando non è stata eseguita 1).
Dopo l’intervento, si esegue un trattamento dell’ambliopia che combina correzione refrattiva (lenti a contatto o occhiali) e occlusione dell’occhio sano (cerotto). Nel PFV unilaterale, la compliance al trattamento dell’ambliopia determina la prognosi visiva. Il trattamento deve essere attivo durante il periodo critico dello sviluppo visivo (dalla nascita fino a circa 10 anni di età).
Risultati chirurgici (risultati a 5 anni dello studio PEDIG sulla cataratta PFV)
Per quanto riguarda i risultati visivi complessivi, 4 occhi su 42 (10%, IC 95% 3–23%) hanno raggiunto un’acuità visiva normale per l’età, e il 48% (IC 95% 32–64%) ha raggiunto un’acuità visiva di 20/200 o migliore 1). Il tasso di raggiungimento di 20/200 o migliore negli occhi pseudofachici PFV (23%) era significativamente peggiore rispetto agli occhi pseudofachici non PFV (68%) (OR aggiustato per età=0,14, P=0,005) 1). Gli occhi afachici PFV hanno mostrato risultati simili rispetto agli occhi afachici non PFV (OR aggiustato per età=1,90, P=0,14) 1).
Per tipo, l’acuità visiva mediana nel tipo anteriore era 20/100 (54% con 20/200 o migliore), mentre nel tipo posteriore l’acuità visiva mediana era peggiore, 20/800 (36% con 20/200 o migliore), con una differenza di 4 linee logMAR (P=0,09) 1).
In altri rapporti, lo studio monocentrico di 20 anni di Bata et al. (58 occhi afachici PFV) ha mostrato che il 33% aveva un’acuità visiva di 20/200 o migliore (follow-up medio 6,7 anni) 4), il rapporto di Anteby et al. (30 occhi afachici PFV) ha mostrato il 16,7% con 20/200 o migliore (follow-up medio 8,5 anni) 5), e de Saint Sauveur et al. hanno riportato che su 36 casi di PFV misto grave, solo il 19% aveva un’acuità visiva misurabile e il 61% non aveva percezione luminosa 8).
Altre complicanze maggiori includono l’opacizzazione dell’asse visivo (45% degli occhi pseudofachici PFV, 15% degli occhi afachici), e il tasso cumulativo di chirurgia di chiarificazione dell’asse visivo raggiunge il 40% negli occhi pseudofachici PFV1). Inoltre, il 13% (IC 95% 2–22%) degli occhi afachici PFV necessita di impianto secondario di IOL entro 5 anni 1).
Non esiste un trattamento efficace per la displasia retinica. Nel tipo posteriore, lo sviluppo visivo è spesso improbabile. In caso di microftalmia marcata, si ricorre precocemente all’uso di una protesi oculare per ragioni estetiche. L’uso della protesi oculare contribuisce anche alla crescita orbitaria.
QQuale acuità visiva ci si può aspettare dopo un intervento PFV?
A
Nel tipo anteriore (limitato alla parte posteriore del cristallino), l’acuità visiva mediana a 5 anni dall’intervento è 20/100, con un tasso di raggiungimento di 20/200 o migliore del 54% 1). Tuttavia, solo il 10% di tutti i pazienti raggiunge un’acuità visiva normale per l’età. Nel tipo posteriore, l’acuità visiva mediana è 20/800, che è scarsa 1). La prognosi visiva è fortemente influenzata dal tipo di PFV, dalla presenza di lesioni del segmento posteriore, dall’età chirurgica e dalla compliance al trattamento dell’ambliopia.
QQual è la complicanza più importante da monitorare dopo un intervento PFV?
A
Gli eventi avversi correlati al glaucoma sono i più frequenti, raggiungendo un’incidenza cumulativa a 5 anni del 24% negli occhi afachici con PFV1). L’opacità dell’asse visivo è anche una complicanza importante, che si verifica nel 45% degli occhi pseudofachici 1). La vitrectomia anteriore al momento del primo intervento è importante per prevenire l’opacità dell’asse visivo (18% con vs 60% senza) 1). Dopo l’intervento, sono necessari una gestione a lungo termine della pressione intraoculare, una valutazione dell’asse visivo e una correzione refrattiva regolari.
6. Fisiopatologia e meccanismi dettagliati di insorgenza
Alla 5a-6a settimana di gestazione, le cellule mesenchimali entrano nella cavità vitrea attraverso la fessura ottica, formando il sistema vascolare vitreale composto dall’arteria ialoidea e dalla tunica vascolosa del cristallino. Questo sistema vascolare svolge un ruolo cruciale nel fornire ossigeno e nutrienti al cristallino in via di sviluppo e al segmento anteriore dell’occhio.
La 10a settimana di gestazione è il picco dello sviluppo, dopodiché inizia la regressione dalla periferia. La regressione si intensifica dalla 13a alla 15a settimana e si completa quasi alla fine della gestazione. Al momento del parto normale, il sistema vascolare vitreale è quasi assente, sebbene possa rimanere una traccia come macchia di Mittendorf (un piccolo punto bianco sulla superficie posteriore del cristallino).
Nel PFV, si verifica una regressione incompleta dei vasi vitreali, portando a uno sviluppo anomalo del tessuto fibroproliferativo perivascolare. Il tessuto mesenchimale perivascolare residuo prolifera e si fibrotizza, dando luogo a varie manifestazioni cliniche.
Le conseguenze specifiche sono le seguenti:
Opacità del cristallino (cataratta sottocapsulare posteriore, lenticono posteriore): adesione del tessuto fibroso residuo alla superficie posteriore del cristallino
Microftalmia: disturbo dello sviluppo dell’intero bulbo oculare dovuto a un’anomalia dello sviluppo del tessuto perivascolare
Displasia retinica: nella forma posteriore, displasia retinica dovuta a trazione e ischemia
Allungamento dei processi ciliari e ipotonia: trazione da parte di cordoni fibrosi
Pieghe retiniche: deformazione dovuta alla trazione del tessuto patologico posteriore
La ragione per cui è unilaterale e non ereditaria è che la regressione dei vasi vitreali fetali è controllata da segnali locali di angiogenesi e regressione, e si ipotizza che un’alterazione di questo meccanismo di controllo si verifichi solo in un occhio. Tuttavia, il meccanismo molecolare preciso non è ancora chiaro.
Il gene specifico responsabile della PFV non è stato ancora identificato, ma in modelli animali è stato dimostrato che mutazioni nei geni correlati alla via di segnalazione Wnt, come FZD4, LRP5 e NDP, mostrano un fenotipo simile alla PFV (persistenza del vitreo primitivo). Questi sono anche geni causali per FEVR e malattia di Norrie, suggerendo una continuità genetica tra PFV e malattie correlate. Anche l’applicazione clinica dei test panel di sequenziamento di nuova generazione (NGS) per la diagnosi differenziale della PFV sta progredendo.
Si stanno tentando approcci alla PFV posteriore mediante vitrectomia a microincisione (MIVS) di calibro 25 e 27. La chirurgia mini-invasiva nei neonati e nei lattanti ha il vantaggio di minimizzare il trauma e sopprimere l’infiammazione postoperatoria11).
Il confronto dei risultati a lungo termine tra l’impianto primario della IOL nell’infanzia e l’impianto secondario dopo gestione dell’afachia è un tema importante nella ricerca sulla PFV. I dati di follow-up esteso dello studio PEDIG dovrebbero portare all’istituzione di criteri specifici per la PFV per l’impianto della IOL6)7).
La valutazione non invasiva della PFV mediante fotocamera del fondo a grandangolo e OCT del segmento anteriore è progredita. L’OCT del segmento anteriore è particolarmente utile per la valutazione morfologica del lenticono posteriore e della cataratta sottocapsulare posteriore, nonché per la pianificazione preoperatoria.
Haider KM, Repka MX, Sutherland DR, Hatt SR, Fallaha N, Kraker RT, et al. Outcomes and Complications 5 Years After Surgery for Pediatric Cataract Associated With Persistent Fetal Vasculature. American journal of ophthalmology. 2024;260:30-36. doi:10.1016/j.ajo.2023.11.002. PMID:37939986; PMCID:PMC11005992.
Wilson ME, Trivedi RH, Morrison DG, Lambert SR, Buckley EG, Plager DA, et al. The Infant Aphakia Treatment Study: evaluation of cataract morphology in eyes with monocular cataracts. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2011;15(5):421-6. doi:10.1016/j.jaapos.2011.05.016. PMID:22108352; PMCID:PMC3345197.
Solebo AL, Russell-Eggitt I, Cumberland P, Rahi JS. Congenital cataract associated with persistent fetal vasculature: findings from IoLunder2. Eye (London, England). 2016;30(9):1204-9. doi:10.1038/eye.2016.159. PMID:27472205; PMCID:PMC5023809.
Bata BM, Chiu HH, Mireskandari K, Ali A, Lam WC, Wan MJ. Long-term visual and anatomic outcomes following early surgery for persistent fetal vasculature: a single-center, 20-year review. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2019;23(6):327.e1-327.e5. doi:10.1016/j.jaapos.2019.07.009. PMID:31629823.
Anteby I, Cohen E, Karshai I, BenEzra D. Unilateral persistent hyperplastic primary vitreous: course and outcome. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2002;6(2):92-9. doi:10.1067/mpa.2002.121324. PMID:11997805.
Repka MX, Dean TW, Lazar EL, Yen KG, Lenhart PD, Freedman SF, et al. Cataract Surgery in Children from Birth to Less than 13 Years of Age: Baseline Characteristics of the Cohort. Ophthalmology. 2016;123(12):2462-2473. doi:10.1016/j.ophtha.2016.09.003. PMID:27769584; PMCID:PMC5121052.
Repka MX, Dean TW, Kraker RT, Li Z, Yen KG, de Alba Campomanes AG, Young MP, Rahmani B, et al. Visual Acuity and Ophthalmic Outcomes 5 Years After Cataract Surgery Among Children Younger Than 13 Years. JAMA ophthalmology. 2022;140(3):269-276. doi:10.1001/jamaophthalmol.2021.6176. PMID:35142808; PMCID:PMC8832311.
de Saint Sauveur G, Chapron T, Abdelmassih Y, Chehaibou I, Lecler A, Dureau P, et al. Management and Outcomes of Posterior Persistent Fetal Vasculature. Ophthalmology. 2023;130(8):844-853. doi:10.1016/j.ophtha.2023.03.027. PMID:37044159.
Khandwala N, Besirli C, Bohnsack BL.. Outcomes and surgical management of persistent fetal vasculature. BMJ Open Ophthalmol. 2021;6(1):e000656. doi:10.1136/bmjophth-2020-000656. PMID:34013048; PMCID:PMC8094357.
Hunt A, Rowe N, Lam A, Martin F.. Outcomes in persistent hyperplastic primary vitreous. Br J Ophthalmol. 2005;89(7):859-863. doi:10.1136/bjo.2004.053595. PMID:15965167; PMCID:PMC1772745.
Bata BM, Khalili S, Ali A, Wan MJ, Mireskandari K. Late surgery for unilateral persistent fetal vasculature: long-term visual and anatomic outcomes. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2022;26(6):296.e1-296.e5. doi:10.1016/j.jaapos.2022.09.005. PMID:36265751.
Goldberg MF. Persistent fetal vasculature (PFV): an integrated interpretation of signs and symptoms associated with persistent hyperplastic primary vitreous (PHPV). LIV Edward Jackson Memorial Lecture. American journal of ophthalmology. 1997;124(5):587-626. doi:10.1016/s0002-9394(14)70899-2. PMID:9372715.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.