Die persistierende fetale Vaskulatur (PFV) ist eine angeborene Augenerkrankung, die durch eine unvollständige Rückbildung des embryonalen Glaskörpergefäßsystems entsteht. Die frühere Bezeichnung war persistierender hyperplastischer primärer Glaskörper (PHPV). 1997 schlug Goldberg den Begriff PFV vor, um auch Reste von fibrovaskulärem Gewebe um die Linse herum zu erfassen 12), und dieser Begriff ist heute weitgehend akzeptiert.
Das Glaskörpergefäßsystem besteht aus der von der Papille ausgehenden Hyaloidarterie (hyaloid artery) und der vorderen Tunica vasculosa lentis. Es bildet sich zwischen der 5. und 6. Schwangerschaftswoche, wenn Mesenchymzellen durch die fetale Spalte in die Glaskörperhöhle eindringen, und erreicht seinen Höhepunkt in der 10. Woche. Danach beginnt die Rückbildung ab der 13.–15. Woche von der Peripherie her und endet in der späten Fetalzeit. Bei PFV ist diese Rückbildung unvollständig, was zu Entwicklungsanomalien des perivaskulären Gewebes führt.
Sie gilt als einseitig und nicht erblich, und es wurde kein spezifisches Gen als Ursache identifiziert. Typische Fälle zeigen sich mit Leukokorie und Mikrophthalmus, können aber auch durch Strabismus oder Amblyopie diagnostiziert werden.
PFV tritt bei etwa 20 % der Säuglings- und Kinderkatarakte auf 2)3). Im Kataraktregister der Pediatric Eye Disease Investigator Group (PEDIG) waren 64 von 994 Fällen (6,4 %) PFV-assoziierte Katarakte 1), von denen 75 % als Aphake operiert wurden (medianes Operationsalter 2 Monate) und 25 % als Pseudophake (medianes Operationsalter 29 Monate) 1). Bei 28 % fanden sich Hinterabschnittsanomalien (Glaskörper-, Netzhaut- oder Sehnervanomalien) 1), und nur 4–6 % hatten eine Familienanamnese 1).
QWas ist der Unterschied zwischen PFV und PHPV?
A
Im Wesentlichen handelt es sich um einen Unterschied zwischen alter und neuer Bezeichnung derselben Erkrankung. PHPV ist die alte Bezeichnung, die sich auf die Hyperplasie und Persistenz der hyaloiden Gefäße (primärer Glaskörper) konzentrierte. 1997 schlug Goldberg den Begriff PFV (persistent fetal vasculature) vor, um auch die Persistenz von fibrovaskulärem Gewebe um die Linse herum zu umfassen. PFV kann ein breiteres Läsionsspektrum abbilden und ist heute die Standardbezeichnung.
QTritt PFV auf beiden Augen auf?
A
PFV ist fast immer unilateral und gilt als nicht erblich. In der PEDIG-Studie wurden nur 3 Fälle von bilateralem PFV ausgeschlossen 1). Bei Bilateralität ist eine Abgrenzung zur familiären exsudativen Vitreoretinopathie (FEVR) oder zum Morbus Norrie erforderlich, und eine Abklärung einschließlich Gentests wird empfohlen.
Egbu E The Outcome of Manual Small Incision Cataract Surgery and Anterior Vitrectomy for Persistent Fetal Vasculature in an 18-Year-Old Woman: A One-Year Follow-Up. Cureus. 2020. Figure 1. PMCID: PMC7584328. License: CC BY.
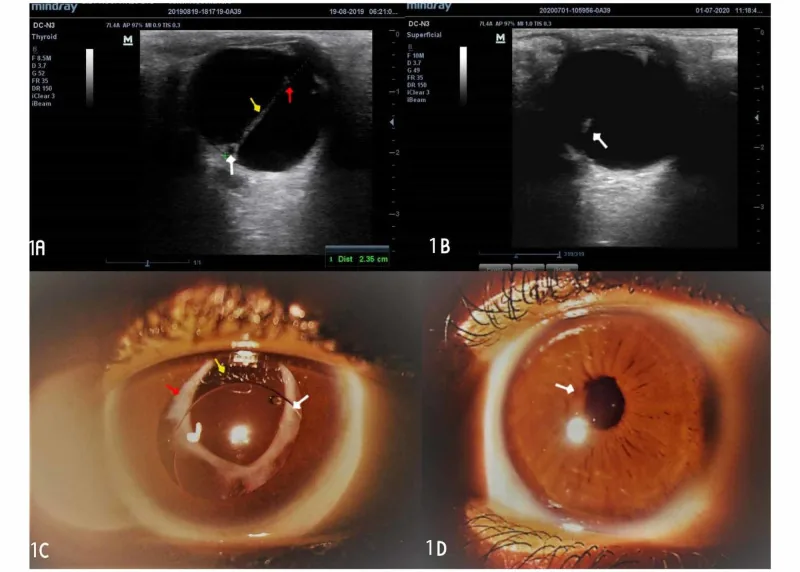
Der Augenultraschall zeigt eine hyperechogene strangförmige Läsion, die durch den Glaskörper verläuft, und das Vorderabschnittsfoto zeigt unter Mydriasis weißes fibrovaskuläres Gewebe hinter der Pupille. Diese Befunde, die die für PFV charakteristische retrolentale fibrovaskuläre Membran und Glaskörperstränge darstellen, sind für den Abschnitt über Hauptsymptome und klinische Befunde geeignet.
PFV wird je nach vorherrschender Lokalisation der Läsionen in drei Typen eingeteilt: anteriorer Typ, posteriorer Typ und gemischter Typ.
Anteriorer Typ
Hauptbefunde : Fibroproliferative Membran auf der Linsenrückfläche (Mittendorf-Fleck), hintere subkapsuläre Katarakt, Lenticonus posterior
Therapieindikation : Operation indiziert, wenn die Läsion auf den hinteren Linsenbereich beschränkt ist und der Fundus normal ist
Prognose : Die beste der drei Typen. Medianer Visus 20/100 (Visus ≥20/200 bei 54%) 1)
Posteriorer Typ
Hauptbefunde : Strang von der Papille in den Glaskörper, Netzhautfalte, Netzhautablösung, Netzhautdysplasie
Therapieindikationen : Bei gutem ERG/VEP ist ein chirurgischer Eingriff in Betracht zu ziehen. Für Netzhautdysplasie gibt es keine wirksame Behandlung.
Prognose : Ungünstig. Medianer Visus 20/800 (Visus ≥20/200 bei 36%)1)
Mischtyp
Hauptbefunde : Mischung aus Befunden des vorderen und hinteren Typs. Häufig mit Mikrophthalmie.
Therapieindikationen : Je nach Ausdehnung der einzelnen Läsionen sorgfältig entscheiden.
Prognose : Bei schwerem Mischtyp haben nur 19% einen messbaren Visus, 61% haben keine Lichtwahrnehmung8)
Detaillierte 7-Typen-Klassifikation in der pädiatrischen Ophthalmologie
Zu den Hauptbefunden gehören einseitige Mikrophthalmie, Trübung der Linsenhinterfläche (Mittendorf-Fleck), abnormer Verlauf der Netzhautgefäße, Netzhautdysplasie, verlängerte Ziliarfortsätze und ein Strang, der von der Linsenhinterfläche zur Papille zieht. Der Strang kann zu Traktion führen und über die Verlängerung der Ziliarfortsätze eine Hypotonie verursachen. In der Nähe der Papille kann die Netzhaut in das Stranggewebe einbezogen sein. Der Schweregrad variiert je nach Rückbildungsgrad der Glaskörpergefäße.
In der PEDIG-Studie wiesen 28 % (18/64 Augen) der PFV-Katarakte Hinterabschnittsanomalien auf 1), und 46 % der pseudophaken Augen hatten eine Hinterabschnittserkrankung (altersadjustierte OR 4,47 im Vergleich zu 28 % der aphaken Augen) 1). Bei 96 % der aphaken PFV-Augen wurde zum Zeitpunkt der Kataraktoperation eine vordere Vitrektomie durchgeführt 1).
Bei der normalen Entwicklung des Glaskörpergefäßsystems dringen mesenchymale Zellen in der 5. bis 6. Schwangerschaftswoche durch die embryonale Spalte in die Glaskörperhöhle ein und bilden das Glaskörpergefäßsystem. Es erreicht seinen Höhepunkt in der 10. Woche, danach beginnt die Rückbildung von der Peripherie her. Konkret beginnt die Rückbildung in der 13. bis 15. Woche und klingt im späten Fetalstadium ab.
Bei PFV kommt es zu einer unvollständigen Rückbildung der Glaskörpergefäße, was zu Entwicklungsanomalien des perivaskulären Gewebes führt. Dies führt zu Linsentrübung, Netzhautdysplasie und Mikrophthalmie. Darüber hinaus verursacht die Traktion des Restgewebes eine Verlängerung der Ziliarfortsätze und Netzhautfalten.
PFV gilt als einseitig und nicht erblich, und es wurde kein spezifisches Gen als Ursache identifiziert. Nur 4–6 % haben eine Familienanamnese 1), die meisten sind sporadisch. Bei beidseitigem Auftreten ist eine genetische Differenzialdiagnose zur FEVR (NDP-, LRP5-, FZD4-Genmutationen) oder zum Morbus Norrie (NDP-Genmutation) wichtig.
Es wurde kein eindeutiger Zusammenhang mit niedrigem Geburtsgewicht oder Frühgeburtlichkeit nachgewiesen, aber schwere Glaskörpergefäßreste können selten ähnliche Befunde wie eine Frühgeborenenretinopathie aufweisen.
Ultraschall (B-Scan): Darstellung von Strängen hinter der Linse. Nützlich zur Beurteilung von Mikrophthalmus und des Glaskörperraums. Besonders wichtig, wenn die durchsichtigen Medien getrübt sind, da der hintere Pol dann nicht direkt sichtbar ist.
CT: Nachweis intraokularer Verkalkungen. Retinoblastom weist Verkalkungen auf, PFV nicht, daher die nützlichste Untersuchung zur Unterscheidung der beiden Erkrankungen.
MRT: Hervorragend zur Beurteilung von Strängen, Anomalien des hinteren Augenabschnitts und Infiltration des Sehnervs.
ERG und VEP (Elektroretinogramm und visuell evozierte Potenziale): Beurteilung der Netzhautfunktion. Ermöglicht die präoperative Bewertung der Sehfunktion und dient als Grundlage für die Entscheidung über einen chirurgischen Eingriff.
Die Abgrenzung zu Erkrankungen, die eine Leukokorie verursachen, ist von größter Bedeutung. Insbesondere die Unterscheidung vom Retinoblastom hat direkte Auswirkungen auf die Lebensprognose.
Abgrenzung zum Retinoblastom
Erkrankung mit höchster Priorität auszuschließen
PFV geht in der Regel mit Mikrophthalmus einher, Retinoblastom nicht. Die CT zum Nachweis intraokularer Verkalkungen ist hilfreich; Verkalkungen sind charakteristisch für Retinoblastom und fehlen bei PFV.
Abgrenzung zur FEVR und zum Norrie-Syndrom
Wichtig bei beidseitigem Befall
Bei beidseitiger totaler Netzhautablösung oder retrolentaler fibröser Proliferation ist eine Abgrenzung zur FEVR oder zum Norrie-Syndrom erforderlich. Familienanamnese und Suche nach genetischen Anomalien sind hilfreich. Die Verwendung von Next-Generation-Sequencing (NGS)-Panel-Tests ist nützlich.
Erkrankung
Seitenverteilung
Mikrophthalmus
Vererbbarkeit
CT-Verkalkung
Charakteristische Befunde
PFV
Fast immer einseitig
Ja
Nicht erblich
Nein
Retrolentale fibrovaskuläre Masse, Membran zur Papille
QWelche Erkrankung sollte bei Leukokorie am dringendsten ausgeschlossen werden?
A
Das Retinoblastom. Es ist der häufigste intraokuläre maligne Tumor bei Kindern und hat direkten Einfluss auf die Lebensprognose, daher ist ein schneller Ausschluss erforderlich. Die PFV geht oft mit einer Mikrophthalmie einher, während beim Retinoblastom normalerweise keine Mikrophthalmie vorliegt. Die CT-Untersuchung auf intraokuläre Verkalkungen ist der wichtigste differenzialdiagnostische Schritt. Bei bestätigten Verkalkungen ist ein Retinoblastom stark zu vermuten und eine Überweisung an ein spezialisiertes Zentrum erforderlich.
Die Trübung der Linse wird wie bei Katarakt behandelt. Ist sie auf den hinteren Linsenbereich beschränkt und der Fundus unauffällig, erfolgen Lensektomie und Exzision der fibroproliferativen Membran analog zur angeborenen Katarakt. Wenn die strangförmigen Gebilde hinter der Linse exzentrisch sind und die Sehachse nicht betreffen, ist eine Operation nicht zwingend erforderlich. Bei Beteiligung des Fundus ist eine Operation in der Regel nicht indiziert. Bei Netzhautablösung oder Traktion ist ein gutes ERG/VEP die Grundlage für einen chirurgischen Eingriff. Bei Pupillendeformation oder -verschluss durch Proliferation im vorderen Linsenbereich wird eine Pupilloplastik durchgeführt.
Nach Entfernung der Linse erfolgen Refraktionskorrektur und Amblyopiebehandlung. Die Operationstechnik entspricht der bei angeborener Katarakt. In der PEDIG-Studie wurde bei 96 % der aphaken PFV-Augen zum Zeitpunkt der Kataraktoperation eine vordere Vitrektomie durchgeführt 1). Die Inzidenz einer Visustrübungen betrug 18 %, wenn bei der Erstoperation eine vordere Vitrektomie durchgeführt wurde, gegenüber 60 %, wenn dies nicht der Fall war 1).
Postoperativ wird eine Amblyopiebehandlung durchgeführt, die Refraktionskorrektur (Kontaktlinsen oder Brille) und Okklusion des gesunden Auges (Augenpflaster) kombiniert. Bei einseitigem PFV bestimmt die Compliance der Amblyopiebehandlung die Sehprognose. Die Behandlung sollte während der kritischen Phase der Sehentwicklung (von der Geburt bis etwa zum 10. Lebensjahr) aktiv durchgeführt werden.
Hinsichtlich der Gesamtsehschärfe erreichten 4 von 42 Augen (10 %, 95 %-KI 3–23 %) eine altersentsprechende normale Sehschärfe, und 48 % (95 %-KI 32–64 %) erreichten eine Sehschärfe von 20/200 oder besser 1). Die Rate des Erreichens von 20/200 oder besser bei pseudophaken PFV-Augen (23 %) war signifikant schlechter als bei nicht-PFV-pseudophaken Augen (68 %) (altersadjustierte OR = 0,14, P = 0,005) 1). Aphake PFV-Augen zeigten ähnliche Ergebnisse wie aphake Nicht-PFV-Augen (altersadjustierte OR = 1,90, P = 0,14) 1).
Nach Typ betrug der mediane Visus beim vorderen Typ 20/100 (54 % mit 20/200 oder besser), während der mediane Visus beim hinteren Typ mit 20/800 (36 % mit 20/200 oder besser) schlechter war, mit einem Unterschied von 4 logMAR-Linien (P = 0,09) 1).
In anderen Berichten zeigte die 20-Jahres-Einzelzentrumsstudie von Bata et al. (58 aphake PFV-Augen), dass 33 % einen Visus von 20/200 oder besser erreichten (mittlere Nachbeobachtung 6,7 Jahre) 4), der Bericht von Anteby et al. (30 aphake PFV-Augen) zeigte 16,7 % mit 20/200 oder besser (mittlere Nachbeobachtung 8,5 Jahre) 5), und de Saint Sauveur et al. berichteten bei 36 schweren gemischten PFV-Fällen, dass nur 19 % einen messbaren Visus hatten und 61 % keine Lichtwahrnehmung 8).
Weitere Hauptkomplikationen sind die Visustrübungen (45 % der pseudophaken PFV-Augen, 15 % der aphaken Augen), und die kumulative Inzidenz einer Visusklärungsoperation erreicht bei pseudophaken PFV-Augen 40 % 1). Darüber hinaus benötigen 13 % (95 %-KI 2–22 %) der aphaken PFV-Augen innerhalb von 5 Jahren eine sekundäre IOL-Implantation 1).
Für die Netzhautdysplasie gibt es keine wirksame Behandlung. Beim hinteren Augentyp ist die Sehentwicklung oft nicht zu erwarten. Bei ausgeprägter Mikrophthalmie wird aus kosmetischen Gründen frühzeitig eine Augenprothese getragen. Das Tragen einer Augenprothese fördert auch das Orbitawachstum.
QWelche Sehschärfe kann nach einer PFV-Operation erwartet werden?
A
Beim vorderen Typ (auf die hintere Linsenregion beschränkt) beträgt der mediane Visus 5 Jahre postoperativ 20/100, mit einer Rate von 54 % für 20/200 oder besser 1). Allerdings erreichen nur 10 % aller Patienten eine altersentsprechende normale Sehschärfe. Beim hinteren Typ beträgt der mediane Visus 20/800, was schlecht ist 1). Die Sehprognose wird stark beeinflusst durch den PFV-Typ, das Vorhandensein von hinteren Segmentveränderungen, das Operationsalter und die Compliance der Amblyopiebehandlung.
QWelche Komplikation ist nach einer PFV-Operation am wichtigsten zu beachten?
A
Glaukombedingte unerwünschte Ereignisse sind am häufigsten und erreichen eine kumulative 5-Jahres-Inzidenz von 24 % bei aphaken PFV-Augen 1). Die Trübung der Sehachse ist ebenfalls eine wichtige Komplikation und tritt bei 45 % der pseudophaken Augen auf 1). Die vordere Vitrektomie während der ersten Operation ist wichtig zur Prävention der Sehachsentrübung (18 % mit vs. 60 % ohne) 1). Auch nach der Operation sind eine langfristige regelmäßige Augeninnendruckkontrolle, Beurteilung der Sehachse und Refraktionskorrektur erforderlich.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
In der 5. bis 6. Schwangerschaftswoche dringen mesenchymale Zellen durch die Augenbecherspalte in die Glaskörperhöhle ein und bilden das Glaskörpergefäßsystem, bestehend aus der Hyaloidarterie und der Tunica vasculosa lentis. Dieses Gefäßsystem spielt eine wichtige Rolle bei der Sauerstoff- und Nährstoffversorgung der sich entwickelnden Linse und des vorderen Augenabschnitts.
Die 10. Schwangerschaftswoche ist der Höhepunkt der Entwicklung, danach beginnt die Rückbildung von der Peripherie aus. Die Rückbildung wird von der 13. bis 15. Schwangerschaftswoche intensiviert und ist im späten Fetalstadium fast vollständig abgeschlossen. Bei der normalen Geburt ist das Glaskörpergefäßsystem fast nicht vorhanden, obwohl ein Rest als Mittendorf-Fleck (kleiner weißer Punkt auf der Linsenrückfläche) verbleiben kann.
Beim PFV kommt es zu einer unvollständigen Rückbildung der Glaskörpergefäße, was zu einer abnormalen Entwicklung des perivaskulären fibroproliferativen Gewebes führt. Das verbliebene perivaskuläre mesenchymale Gewebe proliferiert und fibrosiert, was zu verschiedenen klinischen Befunden führt.
Konkrete Folgen sind:
Linsentrübung (hintere subkapsuläre Katarakt, hinterer Lentikonus): Anhaftung von Restfasergewebe an der Linsenrückfläche
Mikrophthalmus: Entwicklungsstörung des gesamten Auges aufgrund einer abnormalen Entwicklung des perivaskulären Gewebes
Netzhautdysplasie: Bei der hinteren Form Dysplasie der Netzhaut durch Traktion und Ischämie
Verlängerung der Ziliarfortsätze und Hypotonie: Traktion durch strangförmige Gebilde
Netzhautfalten: Verformung durch Traktion des hinteren pathologischen Gewebes
Der Grund für die Einseitigkeit und Nichtvererbbarkeit ist, dass die Rückbildung der fetalen Glaskörpergefäße durch lokale Angiogenese- und Rückbildungssignale gesteuert wird, und es wird angenommen, dass eine Störung dieses Kontrollmechanismus nur in einem Auge auftritt. Der genaue molekulare Mechanismus ist jedoch noch ungeklärt.
Das spezifische verursachende Gen der PFV wurde noch nicht identifiziert, aber in Tiermodellen wurde gezeigt, dass Mutationen in Genen des Wnt-Signalwegs wie FZD4, LRP5 und NDP einen PFV-ähnlichen Phänotyp (Persistenz des Glaskörpergefäßes) aufweisen. Diese sind auch ursächliche Gene für FEVR und Morbus Norrie, was auf eine genetische Kontinuität zwischen PFV und verwandten Erkrankungen hindeutet. Die klinische Anwendung von Next-Generation-Sequencing (NGS)-Panel-Tests zur Differenzialdiagnose der PFV schreitet ebenfalls voran.
Ansätze zur posterioren PFV mittels 25- und 27-Gauge-Mikroinzisionsvitrektomie (MIVS) werden erprobt. Die minimalinvasive Chirurgie bei Neugeborenen und Säuglingen hat den Vorteil, das Trauma zu minimieren und die postoperative Entzündung zu unterdrücken11).
Der Vergleich der Langzeitergebnisse zwischen primärer IOL-Implantation im Säuglingsalter und sekundärer IOL-Implantation nach aphaker Behandlung ist ein wichtiges Thema der PFV-Forschung. Verlängerte Nachbeobachtungsdaten der PEDIG-Studie lassen die Etablierung PFV-spezifischer Kriterien für die IOL-Implantation erwarten6)7).
Die nicht-invasive Beurteilung der PFV mittels Weitwinkel-Funduskamera und Vorderabschnitts-OCT hat Fortschritte gemacht. Insbesondere das Vorderabschnitts-OCT ist nützlich für die morphologische Beurteilung des hinteren Lentikonus und der hinteren subkapsulären Katarakt sowie für die präoperative Planung.
Haider KM, Repka MX, Sutherland DR, Hatt SR, Fallaha N, Kraker RT, et al. Outcomes and Complications 5 Years After Surgery for Pediatric Cataract Associated With Persistent Fetal Vasculature. American journal of ophthalmology. 2024;260:30-36. doi:10.1016/j.ajo.2023.11.002. PMID:37939986; PMCID:PMC11005992.
Wilson ME, Trivedi RH, Morrison DG, Lambert SR, Buckley EG, Plager DA, et al. The Infant Aphakia Treatment Study: evaluation of cataract morphology in eyes with monocular cataracts. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2011;15(5):421-6. doi:10.1016/j.jaapos.2011.05.016. PMID:22108352; PMCID:PMC3345197.
Solebo AL, Russell-Eggitt I, Cumberland P, Rahi JS. Congenital cataract associated with persistent fetal vasculature: findings from IoLunder2. Eye (London, England). 2016;30(9):1204-9. doi:10.1038/eye.2016.159. PMID:27472205; PMCID:PMC5023809.
Bata BM, Chiu HH, Mireskandari K, Ali A, Lam WC, Wan MJ. Long-term visual and anatomic outcomes following early surgery for persistent fetal vasculature: a single-center, 20-year review. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2019;23(6):327.e1-327.e5. doi:10.1016/j.jaapos.2019.07.009. PMID:31629823.
Anteby I, Cohen E, Karshai I, BenEzra D. Unilateral persistent hyperplastic primary vitreous: course and outcome. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2002;6(2):92-9. doi:10.1067/mpa.2002.121324. PMID:11997805.
Repka MX, Dean TW, Lazar EL, Yen KG, Lenhart PD, Freedman SF, et al. Cataract Surgery in Children from Birth to Less than 13 Years of Age: Baseline Characteristics of the Cohort. Ophthalmology. 2016;123(12):2462-2473. doi:10.1016/j.ophtha.2016.09.003. PMID:27769584; PMCID:PMC5121052.
Repka MX, Dean TW, Kraker RT, Li Z, Yen KG, de Alba Campomanes AG, Young MP, Rahmani B, et al. Visual Acuity and Ophthalmic Outcomes 5 Years After Cataract Surgery Among Children Younger Than 13 Years. JAMA ophthalmology. 2022;140(3):269-276. doi:10.1001/jamaophthalmol.2021.6176. PMID:35142808; PMCID:PMC8832311.
de Saint Sauveur G, Chapron T, Abdelmassih Y, Chehaibou I, Lecler A, Dureau P, et al. Management and Outcomes of Posterior Persistent Fetal Vasculature. Ophthalmology. 2023;130(8):844-853. doi:10.1016/j.ophtha.2023.03.027. PMID:37044159.
Khandwala N, Besirli C, Bohnsack BL.. Outcomes and surgical management of persistent fetal vasculature. BMJ Open Ophthalmol. 2021;6(1):e000656. doi:10.1136/bmjophth-2020-000656. PMID:34013048; PMCID:PMC8094357.
Hunt A, Rowe N, Lam A, Martin F.. Outcomes in persistent hyperplastic primary vitreous. Br J Ophthalmol. 2005;89(7):859-863. doi:10.1136/bjo.2004.053595. PMID:15965167; PMCID:PMC1772745.
Bata BM, Khalili S, Ali A, Wan MJ, Mireskandari K. Late surgery for unilateral persistent fetal vasculature: long-term visual and anatomic outcomes. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2022;26(6):296.e1-296.e5. doi:10.1016/j.jaapos.2022.09.005. PMID:36265751.
Goldberg MF. Persistent fetal vasculature (PFV): an integrated interpretation of signs and symptoms associated with persistent hyperplastic primary vitreous (PHPV). LIV Edward Jackson Memorial Lecture. American journal of ophthalmology. 1997;124(5):587-626. doi:10.1016/s0002-9394(14)70899-2. PMID:9372715.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.