Weiße Pupille (Leukokorie) bedeutet „weiße Pupille“ und leitet sich von den griechischen Wörtern leukos (weiß) und kore (Pupille) ab. Es bezeichnet einen Zustand, bei dem anstelle des normalen roten Reflexes (roter Reflex) ein weißer Lichtreflex bei der Beleuchtung des Augenhintergrunds durch die Pupille beobachtet wird.
Wenn sich irgendwo im optischen Weg von der Hornhaut bis zum hinteren Pol eine Trübung oder abnorme Struktur befindet, wird der rote Reflex aus den Aderhautgefäßen blockiert, was zu einem weißen Pupillenreflex führt. Die Ursachen sind vielfältig und umfassen Tumore, angeborene Anomalien, Gefäßerkrankungen, entzündliche Erkrankungen und Trübungen der brechenden Medien.
Die wichtigste Erkrankung in der Differenzialdiagnose der weißen Pupille ist das Retinoblastom. Das Retinoblastom ist der häufigste intraokulare bösartige Tumor bei Kindern, mit einer geschätzten Inzidenz von 1 pro 17.000 Geburten2). Weiße Pupille und Schielen sind die wichtigsten Erstsymptome2), und eine frühzeitige Erkennung ist entscheidend für die Lebens- und Sehprognose.
Darüber hinaus ist die myelinisierte retinale Nervenfaserschicht (MRNFL) eine angeborene Anomalie, die bei etwa 1 % der Bevölkerung auftritt und manchmal als weiße Pupille entdeckt wird1)3).
QWelche Erkrankung sollte bei weißer Pupille am dringendsten ausgeschlossen werden?
A
Das Retinoblastom. Es ist der häufigste intraokulare bösartige Tumor bei Kindern und lebensbedrohlich. Daher muss bei Vorliegen einer weißen Pupille das Retinoblastom mit höchster Priorität ausgeschlossen werden. Details finden Sie im Abschnitt „Diagnose und Untersuchungsmethoden“.
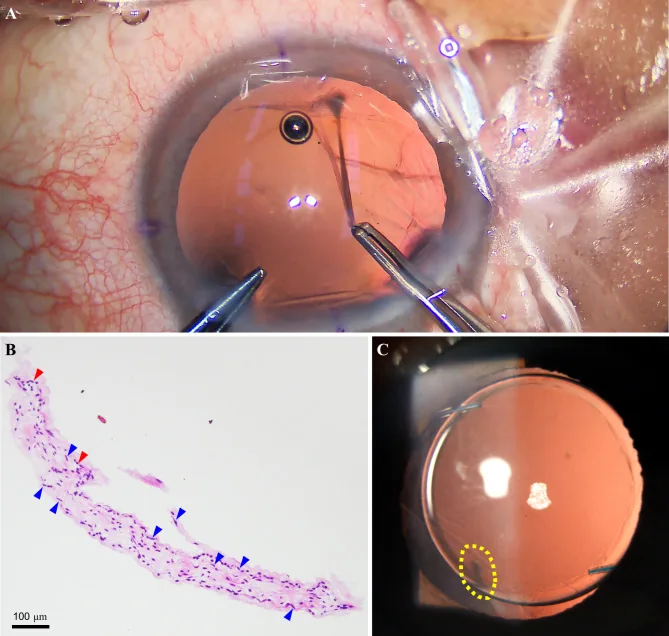
Yu Y, et al. Non-typical persistent hyperplastic primary vitreous: a rare case report and review of the literature. BMC Ophthalmol. 2023. Figure 2. PMCID: PMC10262480. License: CC BY.
Intra- und postoperative Befunde des linken Auges: A zeigt ein fibrovaskuläres Band, das an der hinteren Linsenkapsel anhaftet, B zeigt histologisch Fibroblasten (gelber Pfeil) und Kapillaren (roter Pfeil), C zeigt das normale vordere Augen Segment nach der Operation. Dies entspricht dem fibrovaskulären Band, das im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Ein weißer Pupillenreflex wird in der Regel bei Säuglingen entdeckt, sodass keine subjektiven Symptome berichtet werden. Eltern bemerken ihn häufig in folgenden Situationen:
Weißer Pupillenreflex: Eltern bemerken Auffälligkeiten wie ein weißes Leuchten einer Pupille bei Blitzlichtaufnahmen oder im Dunkeln.
Schielen: Bei Retinoblastom führt ein Tumor in der Makula zu Sehschwäche und Schielen, was zur Entdeckung führt.
Anzeichen von Sehschwäche: Bei Säuglingen können Reiben der Augen, vermindertes Trinken oder seitendifferente Fixation Hinweise sein.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Das normale rote Reflex wird durch einen weißen oder grauweißen Reflex ersetzt. Auch die Asymmetrie des Reflexes zwischen beiden Augen (positiver Bruckner-Reflex) ist ein wichtiger Befund.
QSollte man einen Arzt aufsuchen, wenn ein Auge auf Fotos weiß erscheint?
A
Eine Asymmetrie des roten Reflexes ist eine Indikation für eine augenärztliche Untersuchung. Die American Academy of Pediatrics (AAP) empfiehlt bei allen Neugeborenen, Säuglingen und Kindern eine Untersuchung des roten Reflexes. Ein weißer Reflex oder eine Asymmetrie des Reflexes erfordert eine sofortige Überweisung an einen Augenarzt.
Retinoblastom: Bei Vorliegen einer Keimbahnmutation des RB1-Gens autosomal-dominant vererbt. Die genetische Form macht 35–45 % aller Fälle aus, jedoch haben nur etwa 10 % eine positive Familienanamnese; die meisten sind De-novo-Mutationen 4)
Morbus Coats: Nicht erblich, sporadisch, über 75 % einseitig 6)
ROP: Niedriges Geburtsgewicht, niedriges Gestationsalter und Sauerstoffgabe sind Hauptrisikofaktoren 4)
Nach der Empfehlung der American Academy of Pediatrics (AAP) von 2008 sollte bei allen Neugeborenen, Säuglingen und Kindern vor der Entlassung aus dem Krankenhaus und bei regelmäßigen Vorsorgeuntersuchungen ein Roter Reflex Test durchgeführt werden.
Durchführung: Stellen Sie die Linsenstärke des direkten Ophthalmoskops auf „0“ ein und leuchten Sie aus etwa 45 cm Entfernung in einem abgedunkelten Raum gleichzeitig in beide Augen
Normalbefund: Beide Augen zeigen einen symmetrischen roten Reflex
Abnorme Befunde: Skotome im roten Reflex, deutliche Abschwächung des Reflexes, weißer Reflex, Asymmetrie des Reflexes (Bruckner-Reflex)
Alle oben genannten Anomalien sind Indikationen für eine Überweisung an einen Augenarzt. Die Durchleuchtungsmethode ist nützlich für die Diagnose von Katarakten und auch wichtig für die Beurteilung der Operationsindikation bei Säuglingskatarakten.
Ultraschall (B-Scan): Besonders nützlich, wenn der hintere Pol aufgrund von Trübungen der optischen Medien nicht direkt sichtbar ist. Beim Retinoblastom werden solide Tumoren und innere Verkalkungen nachgewiesen. Bei der Coats-Krankheit zeigt sich keine Verkalkung und eine seröse Netzhautablösung ohne Aderhauttumor 6)
MRT: Überlegen gegenüber CT und Ultraschall zur Unterscheidung von Retinoblastom und Coats-Krankheit6). Das Retinoblastom zeigt in T1-gewichteten Bildern eine ähnliche Signalintensität wie das Hirnparenchym, in T2-gewichteten Bildern eine leicht verminderte Signalintensität und eine Kontrastmittelaufnahme. Bei der Coats-Krankheit zeigt die subretinale Flüssigkeit ein hohes T1- und niedriges T2-Signal ohne Kontrastmittelanreicherung 6). Da etwa 3% der bilateralen Retinoblastome ein trilaterales Retinoblastom entwickeln, wird ein MRT-Screening des Kopfes empfohlen.
Optische Kohärenztomographie (OCT): Bei MRNFL zeigt sich eine hyperreflektive Nervenfaserschicht, auch zur Beurteilung der Makulastruktur nützlich 1)3)
QWie unterscheidet man den Einsatz von Ultraschall und MRT?
A
Die Ultraschalluntersuchung eignet sich hervorragend zur Erkennung von Verkalkungen und kann ambulant schnell durchgeführt werden, sodass sie als initiales Screening geeignet ist. Die MRT ist am besten geeignet, um Retinoblastom von Morbus Coats und persistierendem fetalen Gefäßsystem zu unterscheiden, und ermöglicht auch die Beurteilung einer Infiltration des Sehnervs oder der Aderhaut.
Die Behandlung der Leukokorie variiert stark je nach zugrundeliegender Erkrankung. Im Folgenden werden die Behandlungen für die wichtigsten Ursachen dargestellt.
Retinoblastom
Systemische Chemotherapie: Die Dreifachkombination aus Vincristin 1,5 mg/m², Etoposid 150 mg/m² und Carboplatin 560 mg/m² ist das Standardregime2).
Lokale Therapie: Transpupilläre Thermotherapie (TTT), Laserphotokoagulation und Kryotherapie werden in Kombination mit der Chemotherapie eingesetzt2).
Enukleation: Bei fortgeschrittenen Fällen (z. B. Murphree-Klassifikation Gruppe E) wird eine Enukleation durchgeführt2). Eine Biopsie des intraokularen Tumors wird grundsätzlich nicht durchgeführt, da das Risiko einer extraokularen Tumorzellaussaat besteht.
Coats-Krankheit
Frühstadium (Stadium 1-2): Verschluss erweiterter Gefäße durch Laserphotokoagulation6).
Mittleres Stadium (starke Exsudation): Zerstörung abnormaler Gefäße durch Kryokoagulation6).
Fortgeschrittenes Stadium (mit Netzhautablösung): Netzhautablösungsoperation mittels Vitrektomie usw. Unbehandelt kann es von sekundärem Glaukom bis zur Enukleation kommen6).
MRNFL und Amblyopie
MRNFL selbst: Gutartige angeborene Anomalie, keine Behandlung erforderlich3).
Begleitende Amblyopie: Refraktionskorrektur (Brille oder Kontaktlinsen) und Okklusion (Abdecken des gesunden Auges) sind die Grundlage. Frühzeitige Intervention in der kritischen Phase der Sehentwicklung (bis etwa 10 Jahre) ist wichtig1)3).
Begleitender Strabismus: Bei großem Winkel des Exotropie usw. wird eine Augenmuskeloperation durchgeführt3).
Bei anderen Grunderkrankungen werden bei Frühgeborenenretinopathie (ROP) eine Laserphotokoagulation oder intravitreale Injektion von Anti-VEGF-Medikamenten 4), bei angeborenem Katarakt eine Kataraktoperation und bei persistierendem hyperplastischem primärem Glaskörper (PHPV) eine Linsenektomie und Vitrektomie durchgeführt.
QIst eine Behandlung der Leukokorie durch markhaltige Nervenfasern erforderlich?
A
Markhaltige retinale Nervenfasern (MRNFL) selbst sind eine gutartige angeborene Anomalie und bedürfen keiner Behandlung. Da sie jedoch häufig mit hoher Myopie und Amblyopie einhergehen, sind eine frühzeitige Refraktionskorrektur und Okklusionstherapie für die visuelle Prognose entscheidend. Einzelheiten finden Sie im Abschnitt „Standardbehandlungen“.
6. Pathophysiologie und detaillierter Entstehungsmechanismus
Das Licht des Ophthalmoskops durchdringt Hornhaut, Vorderkammer, Linse und Glaskörper und erreicht die Netzhaut. Die reichlich vorhandenen Blutgefäße in der Aderhaut unter der Netzhaut reflektieren das Licht und werden als roter Reflex beobachtet. Wenn sich in diesem optischen Pfad eine Trübung, ein Tumor oder eine abnormale Struktur befindet, wird der rote Reflex blockiert und es entsteht eine Leukokorie.
Die beidseitige Inaktivierung des Tumorsuppressorgens RB1 (lokalisiert auf Chromosom 13q14) führt zu unkontrollierter Proliferation von Netzhautzellen2)4).
Keimbahnform (heritable form): Eine Mutation in einem Allel liegt in der Keimbahn vor (erster Treffer), nach der Geburt kommt eine somatische Mutation (zweiter Treffer) hinzu. Häufig bilateral4)
Sporadische Form (sporadic form): Beide Allele werden durch somatische Mutationen inaktiviert. Häufig unilateral
Ein weißer Netzhauttumor blockiert direkt den roten Reflex und führt zu einer weißen Pupille.
Es handelt sich um eine nicht-erbliche, sporadische Erkrankung, für die zwei pathogenetische Wege vorgeschlagen wurden6).
Endothel-Degenerationsweg: Die Endothelzellen der Netzhautgefäße degenerieren, wodurch die Blut-Retina-Schranke zusammenbricht. Plasma tritt durch die Gefäßwand aus und führt zu einer Verdickung der Gefäßwand (wurstförmige Gefäße).
Aneurysma-Bildungsweg: Durch abnormale Interaktion zwischen Endothelzellen und Perizyten entstehen Aneurysmen. Die Lipidausscheidung führt zu einer Verdickung der Netzhaut, was zur Zystenbildung und exsudativen Netzhautablösung führt.
Normalerweise findet die Myelinisierung der Nervenfasern nur hinter der Lamina cribrosa statt, und die Nervenfasern in der Netzhaut sind unmyelinisiert. Bei MRNFL dringen aufgrund einer Unreife oder eines Defekts der Lamina cribrosa oligodendrozytenähnliche Zellen in die Netzhaut ein und bilden Myelinscheiden um die Axone der retinalen Ganglienzellen 3). Myelin behindert die Lichtdurchlässigkeit, sodass eine ausgedehnte MRNFL als Leukokorie erkannt wird.
7. Aktuelle Forschung und zukünftige Perspektiven (Forschungsberichte)
Badalova et al. (2025) berichteten in einer retrospektiven Kohortenstudie unter Verwendung des niederländischen nationalen Retinoblastom-Registers (1991–2019), dass alle 28 Fälle von familiärem Retinoblastom, die von Geburt an vollständig gescreent wurden, vor dem Alter von 1 Jahr diagnostiziert wurden (medianes Diagnosealter 18 Tage, Bereich 3–352 Tage). 57,1 % wurden innerhalb des ersten Lebensmonats, 82,1 % innerhalb von 6 Monaten diagnostiziert. In der Gruppe mit unvollständigem Screening (10 Fälle) betrug das mediane Diagnosealter 420 Tage. Basierend auf diesen Ergebnissen wurde das Protokoll überarbeitet, um das Screening für die Niedrigrisikogruppe (geschätztes Risiko <3 %) auf das Alter von 2 Jahren zu verkürzen 7).
Kombination von angeborenen Hirnfehlbildungen und Retinoblastom
Lomi et al. (2025) berichteten über einen Fall eines bilateralen Retinoblastoms bei einem Säugling mit Dandy-Walker-Syndrom (DWS). Sowohl DWS als auch Retinoblastom wurden mit Chromosom-13q-Anomalien in Verbindung gebracht, aber das gleichzeitige Auftreten beider Erkrankungen ist äußerst selten. In einer MRT-Studie von Rodjan et al. an 168 Kindern mit Retinoblastom wurde nur ein Fall einer Dandy-Walker-Variante gefunden. Dies unterstreicht die Bedeutung des Screenings auf intraokuläre Malignome bei Kindern mit angeborenen Hirnfehlbildungen 2).
Es wird diskutiert, ob eine In-vitro-Fertilisation (IVF) über epigenetische Anomalien das Risiko für die Entwicklung eines Retinoblastoms erhöhen könnte. Eine niederländische Studie deutete auf eine erhöhte Inzidenz von Retinoblastomen nach IVF hin, aber mehrere große epidemiologische Studien fanden keinen signifikanten Zusammenhang, sodass die Schlussfolgerung noch nicht endgültig ist 4).
Die Trias aus myelinisierten retinalen Nervenfasern (MRNFL), Myopie und Amblyopie wird als Straatsma-Syndrom bezeichnet. Als Faktoren, die mit der visuellen Prognose zusammenhängen, wurden der MRNFL-Typ (Typ 1: nur oberer Bogen, Typ 2: oberer und unterer Bogen, Typ 3: nicht mit der Papille verbunden), der Grad der Anisometropie und die Struktur der ellipsoiden Zone der Makula im OCT beschrieben. Typ 2 hat die schlechteste Prognose und spricht auch schlecht auf die Amblyopiebehandlung an 1).
Altamirano F, Gonzalez E, Oke I.. Leukocoria in a 4-year-old boy. Digit J Ophthalmol. 2024;30(2):42-44. doi:10.5693/djo.02.2024.03.005. PMID:38962670; PMCID:PMC11218840.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Alghofaili RS, Almesfer SA.. Bilateral Retinoblastoma Presenting in an in vitro Fertilization Infant with Retinopathy of Prematurity. Case Rep Ophthalmol. 2021;12(1):306-310. doi:10.1159/000513181. PMID:34054475; PMCID:PMC8138227.
Bafna RK, Mahalingam K, Bansal B. Hyperoleon masquerading as leukocoria. BMJ case reports. 2021;14(9). doi:10.1136/bcr-2021-246135. PMID:34544725; PMCID:PMC8454427.
Laasri K, El Houss S, Halfi IM, Kettani NE, Fikri M, Jiddane M, et al. Coats’ syndrome: A rare cause of infant leukocoria to keep in mind. Radiology case reports. 2024;19(1):7-11. doi:10.1016/j.radcr.2023.09.046. PMID:37881471; PMCID:PMC10594555.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At What Age Could Screening for Familial Retinoblastoma Be Stopped? Ophthalmology. 2025;132:1152-1160.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.