
Kolobom der Sehnervenpapille
Auf einen Blick: Wichtige Punkte
Abschnitt betitelt „Auf einen Blick: Wichtige Punkte“1. Was ist ein Papillenkolobom?
Abschnitt betitelt „1. Was ist ein Papillenkolobom?“Das Papillenkolobom ist eine angeborene Anomalie, die durch eine abnorme Vergrößerung der Papille und eine scharf begrenzte weiße Vertiefung gekennzeichnet ist. Es entsteht durch einen unvollständigen Verschluss der Augenbecherspalte (embryonale Spalte), die sich normalerweise in der 7. Schwangerschaftswoche schließt. Die Netzhautgefäße entspringen nicht an einer einzigen Stelle, sondern an verschiedenen Stellen am Rand oder innerhalb der Vertiefung.
Wenn der unvollständige Verschluss der Augenbecherspalte auf den hinteren Teil (Sehnervseite) beschränkt ist, entsteht ein Sehnervenkolobom. Bei einem ausgedehnteren unvollständigen Verschluss von vorne nach hinten bildet sich ein Spektrum vom Iris- bis zum Aderhautkolobom. Im gesamten Kolobomspektrum entspricht das Papillenkolobom dem hinteren Ende des unvollständigen Verschlusses der Augenbecherspalte und ist Teil des kontinuierlichen Spektrums mit dem Iriskolobom (vorderes Ende), es existiert jedoch auch eine isolierte lokalisierte Form der Papille.
Der ICD-10-Code ist H47.319 (Sehnerv).
Die Abgrenzung zum Morning-Glory-Syndrom ist wichtig. Beim Morning-Glory-Syndrom findet sich im Zentrum der Papille ein gliales Proliferationsgewebe, und die Gefäße verlaufen radial. Beim Papillenkolobom gibt es keine gliale Proliferation, die Vertiefung ist nach unten betont, und die Gefäße entspringen an verschiedenen Stellen am Rand oder innerhalb der Vertiefung, was eine Unterscheidung ermöglicht.
Das Papillenkolobom zeigt eine nach unten betonte, scharf begrenzte weiße Vertiefung, und die Gefäße entspringen an verschiedenen Stellen am Rand oder innerhalb der Vertiefung. Es gibt kein gliales Proliferationsgewebe. Beim Morning-Glory-Syndrom befindet sich im Zentrum der Papille ein gliales Proliferationsgewebe, und die Gefäße strahlen radial vom Papillenrand aus. Beide sind angeborene Anomalien der Papille, können aber durch den Fundusbefund unterschieden werden.
2. Hauptsymptome und klinische Befunde
Abschnitt betitelt „2. Hauptsymptome und klinische Befunde“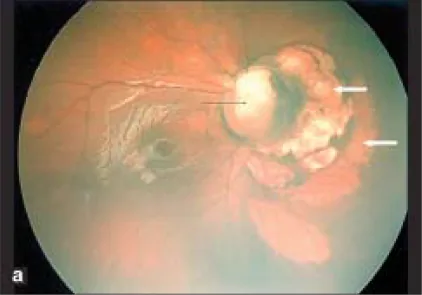
Subjektive Symptome
Abschnitt betitelt „Subjektive Symptome“Die Sehschärfe hängt davon ab, ob das papillomakuläre Bündel in das Kolobom einbezogen ist und in welchem Ausmaß. Sie reicht von über 1,0 bis zu schlechten Fällen, aber aufgrund der Anomalie des Sehnervs ist die Sehschärfe oft auch ohne Makulaschädigung vermindert.
- Sehschärfenminderung: Abhängig vom Ausmaß der Einbeziehung des papillomakulären Bündels. Bei schlechter Sehschärfe kann es zu einem Schielschielen durch Nichtgebrauch kommen.
- Gesichtsfeldausfall: Häufig tritt ein oberer Gesichtsfeldausfall auf, der dem unteren Kolobom der Papille entspricht.
- Schielen: Bei schlechter Sehschärfe kann sich ein Schielen durch Nichtgebrauch entwickeln.
Fundusbefunde
Abschnitt betitelt „Fundusbefunde“Im Fundus zeigt sich ein Defekt der Sehnervenpapille und der unteren Netzhaut und Aderhaut, hauptsächlich nach unten. Es liegen Gefäßverlaufsanomalien vor; die Arteria centralis retinae teilt sich hinter der Papille, sodass zahlreiche Netzhautarterien aus der Papille zu entspringen scheinen. Der obere Rand der Papille bleibt oft erhalten, und selbst wenn die gesamte Papille eingedellt ist, ist die Eindellung typischerweise unten stärker als oben.
Der Papillenbereich ist eingedellt, die Papille fehlt oder ist teilweise defekt, und die umgebende Aderhaut, das retinale Pigmentepithel (RPE) und die Sklera sind verdünnt. Unterhalb der Eindellung des Papillenbereichs finden sich eine Aderhaut-Netzhaut-Atrophie und ein Tigergrund, die auf einen unvollständigen Verschluss der embryonalen Spalte zurückzuführen sind.
Klassifikation
Abschnitt betitelt „Klassifikation“Das Kolobom der Sehnervenpapille wird je nach Ausdehnung wie folgt klassifiziert:
- Isoliertes Papillenkolobom: Verursacht durch einen lokalisierten unvollständigen Verschluss des hinteren Teils der Augenbecherspalte.
- Kombiniertes Aderhaut-Netzhaut-Kolobom: Zeigt einen ausgedehnteren unvollständigen Verschluss der Augenbecherspalte an.
- Kombinierter Iris-Ziliarkörper-Kolobom-Typ : ausgedehnte Form, die vom vorderen bis zum hinteren Ende durchgehend ist.
- Fuchs-Kolobom : milde Form mit einer atrophischen Läsion ähnlich einem Konus unterhalb der Papille. Das Sehvermögen bleibt oft relativ erhalten.
Komplikationen
Abschnitt betitelt „Komplikationen“Augenkomplikationen
Abschnitt betitelt „Augenkomplikationen“- Häufig begleitet von Iris-Kolobom und Aderhaut-Kolobom.
- Bei begleitendem Aderhaut-Kolobom mit großem Einsenkungsbereich kann eine Mikrophthalmie auftreten.
- Seröse Netzhautablösung: kann auch bei isoliertem Sehnervkolobom auftreten.
- Rhegmatogene Netzhautablösung: kann sekundär bei komplexem Aderhaut-Kolobom auftreten.
- Hypotonie durch transsklerale Filtration: Es wurden Fälle von Kammerwasseraustritt durch den Skleradefekt berichtet 7).
3. Ursachen, Epidemiologie und Risikofaktoren
Abschnitt betitelt „3. Ursachen, Epidemiologie und Risikofaktoren“Epidemiologie
Abschnitt betitelt „Epidemiologie“Die Prävalenz wird mit 3–8/100.000 angegeben. Ein- und beidseitige Fälle sind etwa gleich häufig, meist sporadisch, aber oft mit Familienanamnese. Es wurden verschiedene Vererbungsmuster berichtet: autosomal-dominant, autosomal-rezessiv und X-chromosomal 4).
Die gesamte genetische Diagnoserate für Kolobome liegt unter 30 % 5). Viele sporadische Fälle ohne identifizierte Mutation deuten auf Umweltfaktoren und mehrere modifizierende Gene hin.
Entstehungsmechanismus
Abschnitt betitelt „Entstehungsmechanismus“Die embryonale Spalte (Augenbecherspalte) entsteht in der 4. Schwangerschaftswoche ventral bei der Bildung des Augenbechers aus dem Neuroektoderm. Sie ist in der 5. Woche vollständig und beginnt sich ab der 6. Woche zu schließen. Der Verschluss schreitet von der Äquatorregion nach vorne (zur Iris) und nach hinten (zum Sehnerv) fort und ist in der 7. Woche abgeschlossen. Ein lokalisierter hinterer Verschlussdefekt führt zu einem Sehnervkolobom.
Assoziierte Gene
Abschnitt betitelt „Assoziierte Gene“| Gen | Assoziierte Erkrankung | Anmerkungen |
|---|---|---|
| PAX2 | Renales Kolobom-Syndrom (renal coloboma syndrome) | Beteiligt an der ventralen Augenbestimmung und dem Verschluss der embryonalen Spalte1) |
| CHD7 | CHARGE-Syndrom | Chromosom 8 (8q12.2), designierte seltene Krankheit |
| FZD5 | Syndromisches Kolobom + Mikrokornea | Rezeptor des Wnt-Signalwegs2) |
Systemische Komplikationen
Abschnitt betitelt „Systemische Komplikationen“Das Kolobom der Sehnervenpapille kann mit folgenden systemischen Syndromen einhergehen.
- CHARGE-Syndrom : Multiorgan-Fehlbildungssyndrom, benannt nach den Anfangsbuchstaben von Kolobom (C), Herzfehler (H), Choanalatresie (A), Wachstumsstörung (R), Genitalhypoplasie (G) und Ohranomalie (E). Das CHD7-Gen ist das verantwortliche Gen; es ist als seltene Krankheit anerkannt.
- Aicardi-Syndrom : Gekennzeichnet durch Balkenmangel, Epilepsie und geistige Entwicklungsverzögerung. Tritt häufiger bei Mädchen auf. Das Kolobom zeigt sich oft als multiple Lakunen in der Aderhaut und Netzhaut.
- Renales Kolobom-Syndrom : Verursacht durch PAX2-Genmutation. Geht mit Nieren- und Harnwegsfehlbildungen einher. Langzeitkontrolle der Nierenfunktion ist erforderlich. Die Frameshift-Mutation c.76delG in PAX2 wurde in Familien mit fokal-segmentaler Glomerulosklerose (FSGS) identifiziert; das phänotypische Spektrum ist breiter als bisher angenommen 1).
Es kann mit systemischen Syndromen wie CHARGE-Syndrom (Multiorganfehlbildungen durch CHD7-Mutation, seltene Krankheit), Aicardi-Syndrom (Balkenmangel, Epilepsie, weibliche Dominanz) und renalem Kolobom-Syndrom (PAX2-Mutation, Nieren- und Harnwegsfehlbildungen) einhergehen. Bei beidseitigem Befall oder systemischen Befunden werden pädiatrische Konsultation und genetische Beratung empfohlen.
4. Diagnose und Untersuchungsmethoden
Abschnitt betitelt „4. Diagnose und Untersuchungsmethoden“Diagnosemethode
Abschnitt betitelt „Diagnosemethode“Die Diagnose ist allein durch ophthalmoskopische Befunde möglich. Die diagnostischen Hauptmerkmale sind eine scharf begrenzte weiße Vertiefung, die vorwiegend im unteren Papillenbereich liegt, und eine charakteristische Gefäßverlaufsanomalie (zahlreiche Gefäße, die vom Rand oder aus der Vertiefung entspringen). Zur Bestätigung werden Ultraschall, MRT, CT und optische Kohärenztomographie (OCT) eingesetzt.
Obwohl sporadische Fälle häufig sind, kann auch eine Familienanamnese vorliegen, daher ist eine sorgfältige Erhebung der Familienanamnese erforderlich.
Zur Suche nach intrakraniellen Fehlbildungen (wie Balkenmangel) ist eine MRT/CT des Kopfes erforderlich. Eine pädiatrische Konsultation wird durchgeführt, um systemische Komplikationen wie CHARGE-Syndrom oder Aicardi-Syndrom auszuschließen.
Untersuchungen
Abschnitt betitelt „Untersuchungen“| Untersuchung | Ziel |
|---|---|
| Fundusuntersuchung (Mydriasis) | Beurteilung der Papillenmorphologie, des Gefäßverlaufs und einer Netzhautablösung |
| OCT (optische Kohärenztomographie) | Detaillierte Beurteilung der Sehnervenkopf- und Makulastruktur |
| Ultraschalluntersuchung (B-Bild) | Suche nach Netzhautablösung bei schlechter Funduseinsicht |
| Gesichtsfelduntersuchung | Beurteilung des Gesichtsfelddefektmusters (oberer Gesichtsfelddefekt usw.) |
| Kopf-MRT | Suche nach ZNS-Komplikationen wie Balkenagenesie und Enzephalozele |
| Nierenultraschall | Screening auf Nierenkolobom-Syndrom |
| Gentest | Suche nach Mutationen in PAX2, CHD7 usw. (bei bilateralen oder syndromalen Fällen) |
Differenzialdiagnose
Abschnitt betitelt „Differenzialdiagnose“| Differenzialdiagnosen | Abgrenzungsmerkmale |
|---|---|
| Morning-Glory-Syndrom | Gliazellproliferation im Zentrum der Papille, Gefäße radiär. Beim Kolobom überwiegend untere Einbuchtung ohne Gliawucherung |
| Peripapilläres Staphylom | Nach hinten vorgewölbte Sklera um die Papille. Kolobom ist ein Defekt der Papille selbst |
| Papilläres PFV/PHPV (Persistenz der Arteria hyaloidea) | Mit Glaskörperstrang und Netzhautfalten. Fundusbild unterscheidet sich vom Kolobom |
| Megalopapille | Papillendurchmesser vergrößert, aber Form nahezu normal. Keine Exkavation oder Gefäßanomalie 5) |
| Optikushypoplasie | Papille klein (DM/DD-Verhältnis ≥ 3,2). Kolobom zeigt vergrößerte und exkavierte Papille |
| Glaukomatöse Optikusatrophie | Progressive Exkavationsvergrößerung und Augeninnendruckerhöhung. Kolobom ist nicht progressiv mit normalem Druck |
5. Standardbehandlung
Abschnitt betitelt „5. Standardbehandlung“Das Kolobom der Sehnervenpapille ist eine angeborene strukturelle Anomalie, für die es keine kurative Behandlung gibt. Die Behandlung erfolgt symptomatisch, abhängig vom Vorhandensein und der Art der Komplikationen.
Verlaufskontrolle
Abschnitt betitelt „Verlaufskontrolle“Da es sich um eine nicht fortschreitende angeborene Anomalie handelt, wird bei Fehlen von Komplikationen wie einer serösen Netzhautablösung eine regelmäßige Fundusbeobachtung fortgesetzt. Im Kindesalter wird alle sechs Monate bis ein Jahr eine Funduskopie in Mydriasis empfohlen.
Behandlung der serösen Netzhautablösung
Abschnitt betitelt „Behandlung der serösen Netzhautablösung“Es besteht kein Konsens über die Behandlung der serösen Netzhautablösung, und es wurden Fälle von spontaner Rückbildung berichtet. Manchmal wird eine Beobachtung über mehrere Monate durchgeführt. Wenn sich nach der Beobachtung keine Besserung zeigt, wird ein chirurgischer Eingriff in Betracht gezogen.
Es wird angenommen, dass die strukturelle Anomalie der Exkavation den Eintritt von Glaskörperflüssigkeit in den subretinalen Raum ermöglicht, und es wurde auch die Möglichkeit eines Liquorflusses durch eine Verbindung zwischen der Exkavation und dem Subarachnoidalraum vorgeschlagen.
Behandlung der rhegmatogenen Netzhautablösung
Abschnitt betitelt „Behandlung der rhegmatogenen Netzhautablösung“Bei rhegmatogener Netzhautablösung werden eine Vitrektomie und eine Photokoagulation um die Exkavation herum durchgeführt. Die postoperative Sehprognose ist nicht immer günstig.
Eine Netzhautwiederanlegung mit Fibrinkleber wurde bei kolobombedingter Netzhautablösung durchgeführt, wobei Fibrinkleber um den Netzhautriss am Rand des Koloboms aufgetragen wird, um die Adhäsion zu verstärken 3). In einigen Fällen wurde eine Verbesserung des endgültigen Visus auf 20/50 erreicht.
Amblyopiebehandlung
Abschnitt betitelt „Amblyopiebehandlung“Bei schlechtem Visus, insbesondere einseitig, werden eine Refraktionskorrektur und eine Okklusionstherapie (Abdecken des gesunden Auges) durchgeführt. Eine frühzeitige Intervention im Kindesalter ist wichtig. Allerdings ist die Wirksamkeit der Amblyopiebehandlung bei Sehverschlechterung aufgrund einer strukturellen Anomalie des Sehnervs selbst begrenzt.
Systemische Behandlung
Abschnitt betitelt „Systemische Behandlung“- Fälle mit CHARGE-Syndrom: Multidisziplinäre Betreuung mit Herzchirurgie, HNO und Endokrinologie erforderlich.
- Renales Kolobom-Syndrom: Langzeit-Follow-up der Nierenfunktion durchführen.
- Genetische Beratung: Empfohlen bei bilateralen oder syndromalen Fällen.
Eine seröse Netzhautablösung kann sich spontan zurückbilden, daher wird zunächst eine Beobachtung über mehrere Monate durchgeführt. Bei einer rhegmatogenen Ablösung werden eine Vitrektomie und eine Photokoagulation um die Eindellung herum durchgeführt. In letzter Zeit wurde auch eine Verstärkung der Adhäsion mit Fibrinkleber berichtet. Die postoperative Sehprognose ist jedoch nicht immer gut.
6. Pathophysiologie und detaillierte Mechanismen
Abschnitt betitelt „6. Pathophysiologie und detaillierte Mechanismen“Verschlussprozess der embryonalen Spalte
Abschnitt betitelt „Verschlussprozess der embryonalen Spalte“Der Augenbecher bildet sich in der 4. Schwangerschaftswoche aus dem Neuroektoderm. Auf der ventralen Seite des Augenbechers entsteht eine embryonale Spalte (Augenbecherspalte), durch die die Arteria hyaloidea verläuft. Diese Spalte ist in der 5. Woche vollständig und beginnt sich in der 6. Woche zu schließen. Der Verschluss beginnt in der Nähe des Äquators und schreitet nach vorne (zur Iris hin) und nach hinten (zum Sehnerv hin) fort und ist in der 7. Woche abgeschlossen. Ein lokalisierter Verschlussdefekt im hinteren Bereich führt zu einem Sehnervenkolobom.
Am Verschlussprozess ist die epithelial-mesenchymale Transition (EMT) beteiligt. Die Neuroretina-Epithelzellen am Rand der embryonalen Spalte bauen die Basalmembran ab, erwerben mesenchymale Eigenschaften und fusionieren. Eine Störung dieses Prozesses verursacht ein Kolobom6).
Molekulare Mechanismen
Abschnitt betitelt „Molekulare Mechanismen“Das PAX2-Gen ist an der ventralen Determination des Auges beteiligt und spielt eine Rolle beim Verschluss der embryonalen Spalte. PAX2-Mutationen verursachen das renale Kolobom-Syndrom. Eine Frameshift-Mutation c.76delG von PAX2 wurde in einer Familie mit FSGS identifiziert, und der Phänotyp des renalen Kolobom-Syndroms ist breiter als bisher angenommen1).
Das FZD5-Gen kodiert einen Rezeptor des Wnt-Signalwegs. Funktionsverlustmutationen beeinträchtigen die ligandenabhängige Aktivierung des Wnt-Signals, was zu einem unvollständigen Verschluss der embryonalen Spalte und einer Mikrokornea führt. Der Vererbungsmodus ist rezessiv2).
Das CHD7-Gen kodiert einen Chromatin-Remodellierungsfaktor und ist an der Differenzierung und Migration von Neuralleistenzellen beteiligt. Mutationen verursachen das CHARGE-Syndrom.
Mechanismus der serösen Netzhautablösung
Abschnitt betitelt „Mechanismus der serösen Netzhautablösung“Es wird angenommen, dass aufgrund einer strukturellen Anomalie der Exkavation Glaskörperflüssigkeit in den subretinalen Raum fließt. Auch die Möglichkeit eines Liquorflusses durch eine Verbindung zwischen der Exkavation und dem Subarachnoidalraum wurde vorgeschlagen, was als ein Grund für die schwierige Behandlung der serösen Netzhautablösung angesehen wird.
Prognose und Verlauf
Abschnitt betitelt „Prognose und Verlauf“Das Kolobom der Sehnervenpapille ist eine nicht fortschreitende, fixierte angeborene Anomalie. Die Komplikation einer Netzhautablösung ist der Hauptfaktor, der die Sehprognose verschlechtert. Auch nach einer Vitrektomie bei rhegmatogener Netzhautablösung ist die Sehprognose oft nicht gut.
7. Aktuelle Forschung und zukünftige Perspektiven
Abschnitt betitelt „7. Aktuelle Forschung und zukünftige Perspektiven“Erweiterung des phänotypischen Spektrums von PAX2-Mutationen
Abschnitt betitelt „Erweiterung des phänotypischen Spektrums von PAX2-Mutationen“Hu et al. (2024) berichteten über die Identifizierung einer c.76delG-Frameshift-Mutation in PAX2 in einer Familie mit fokal-segmentaler Glomerulosklerose (FSGS) 1). Das Phänotyp des renalen Kolobomsyndroms ist breiter als bisher angenommen, und die Bedeutung des Screenings der Nierenfunktion bei Patienten mit Sehnervenpapillenkolobom wurde erneut hervorgehoben. Es wird vorgeschlagen, die Indikationen für die Nierenfunktionsbewertung bei Patienten mit Kolobom zu erweitern.
FZD5 und Wnt-Signalweg-Anomalien
Abschnitt betitelt „FZD5 und Wnt-Signalweg-Anomalien“Cortes-Gonzalez et al. (2024) berichteten, dass eine homozygote Missense-Mutation (p.M160V) in FZD5 ein symptomatisches okuläres Kolobom und eine Mikrokornea verursacht 2). Die funktionelle Analyse bestätigte, dass die ligandenabhängige Aktivierung des Wnt-Signalwegs in einem rezessiven Vererbungsmuster gestört ist. Die genetische Diagnoserate des Koloboms liegt unter 30 %, und die Identifizierung neuer ursächlicher Gene wird voraussichtlich zur Verbesserung der Diagnose beitragen.
Neue chirurgische Techniken bei Netzhautablösung
Abschnitt betitelt „Neue chirurgische Techniken bei Netzhautablösung“Jain et al. (2024) berichteten über eine Verbesserung der endgültigen Sehschärfe auf 20/50 nach einer Retinopexie mit Fibrinkleber bei kolobombedingter Netzhautablösung 3). Die Technik, Fibrinkleber um den Netzhautriss am Rand des Koloboms aufzutragen, um die Adhäsion zu verstärken, wird als ergänzende Option zur konventionellen Vitrektomie mit Photokoagulation angesehen. Die Sammlung von Fällen und die Überprüfung der Langzeitergebnisse sind zukünftige Aufgaben.
8. Referenzen
Abschnitt betitelt „8. Referenzen“-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.