
Optic Disc Coloboma
Key points at a glance
Section titled “Key points at a glance”1. What is Optic Disc Coloboma?
Section titled “1. What is Optic Disc Coloboma?”Optic disc coloboma is a congenital abnormality characterized by an abnormally enlarged optic disc with a well-defined white depression. It results from incomplete closure of the optic cup fissure (embryonic fissure), which normally closes by the 7th week of gestation. Retinal vessels do not arise from a single point but originate from various locations at the edge or within the depression.
When incomplete closure of the optic cup fissure is limited to the posterior part (optic nerve side), optic disc coloboma occurs. If the incomplete closure is more extensive from anterior to posterior, it forms a spectrum of iris to choroidal coloboma. Within the overall coloboma spectrum, optic disc coloboma corresponds to the posterior end of the incomplete closure, and is part of a continuous spectrum from iris coloboma (anterior end), but an isolated form limited to the optic disc also exists.
The ICD-10 code is H47.319 (optic nerve).
Differentiation from morning glory syndrome is important. In morning glory syndrome, glial proliferation tissue is seen in the center of the disc, and vessels run radially. Optic disc coloboma can be differentiated by the absence of glial proliferation, inferiorly predominant depression, and vessels originating from various sites at the edge or within the depression.
Optic disc coloboma presents with an inferiorly predominant, well-defined white depression, and vessels originate from various sites at the edge or within the depression. No glial proliferation tissue is seen. In morning glory syndrome, glial proliferation tissue is present in the center of the disc, and vessels run radially from the disc margin. Both are congenital optic disc abnormalities, but they can be differentiated by fundus findings.
2. Main Symptoms and Clinical Findings
Section titled “2. Main Symptoms and Clinical Findings”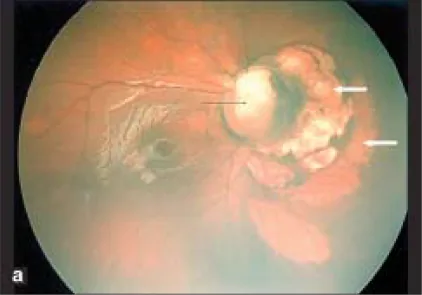
Subjective Symptoms
Section titled “Subjective Symptoms”Visual acuity depends on whether the papillomacular bundle is involved in the coloboma and the extent of involvement. The degree varies from better than 1.0 to poor, but in many cases visual acuity is reduced even without macular involvement due to optic nerve abnormality.
- Decreased visual acuity: Depends on the degree of involvement of the papillomacular bundle. In cases with poor vision, disuse strabismus may occur.
- Visual field defect: Often causes superior visual field defect corresponding to the coloboma inferior to the optic disc.
- Strabismus: Disuse strabismus may develop secondarily in cases with poor vision.
Fundus Findings
Section titled “Fundus Findings”In the fundus, defects of the optic disc and the retinochoroid inferior to the eyeball occur, mainly inferiorly. Vascular abnormalities are present; because the central retinal artery branches posterior to the disc, many retinal arteries appear to emerge from the disc. The superior rim of the disc often remains, and even when the entire disc is depressed, the depression is typically stronger inferiorly compared to superiorly.
The optic disc area is depressed, and the disc is absent or partially defective; the surrounding choroid, retinal pigment epithelium (RPE), and sclera are also thin. Inferior to the depression of the disc area, there is retinochoroidal atrophy and paving-stone appearance due to incomplete closure of the embryonic fissure.
Classification
Section titled “Classification”Optic disc coloboma is classified as follows based on the extent of involvement.
- Isolated optic disc coloboma: Due to localized incomplete closure of the posterior part of the optic cup fissure.
- Combined with retinochoroidal coloboma: Indicates more extensive incomplete closure of the optic cup fissure.
- Iris/ciliary body coloboma combined type: Extensive type continuous from the anterior to the posterior end.
- Fuchs coloboma: A mild type showing an atrophic lesion resembling a conus below the optic disc. Visual acuity is often relatively preserved.
Complications
Section titled “Complications”Ocular complications
Section titled “Ocular complications”- Iris coloboma and choroidal coloboma are often associated.
- When choroidal coloboma is present and the depressed area is large, microphthalmos may occur.
- Serous retinal detachment: Can occur even with optic disc coloboma alone.
- Rhegmatogenous retinal detachment: May occur secondarily in cases with complex retinochoroidal coloboma.
- Hypotony due to transscleral filtration: Cases of aqueous humor leakage from the scleral defect have also been reported7).
3. Causes, Epidemiology, Risk Factors
Section titled “3. Causes, Epidemiology, Risk Factors”Epidemiology
Section titled “Epidemiology”The prevalence is reported to be 3–8 per 100,000. Unilateral and bilateral cases are equally common, and while many cases are sporadic, a family history is often present. Various inheritance patterns including autosomal dominant, autosomal recessive, and X-linked have been reported4).
The overall genetic diagnostic rate for coloboma remains below 30%5). Many sporadic cases without identified gene mutations suggest the involvement of environmental factors and multiple modifier genes.
Pathogenesis
Section titled “Pathogenesis”The embryonic fissure (optic cup fissure) forms ventrally when the optic cup develops from the neuroectoderm at 4 weeks of gestation. It is completed at 5 weeks, and closure begins at 6 weeks. Closure proceeds from near the equator anteriorly (toward the iris) and posteriorly (toward the optic nerve), completing at 7 weeks. Localized failure of posterior closure results in optic disc coloboma.
Related Genes
Section titled “Related Genes”| Gene | Associated Disease | Notes |
|---|---|---|
| PAX2 | Renal coloboma syndrome | Involved in ventral eye determination and embryonic fissure closure1) |
| CHD7 | CHARGE syndrome | Chromosome 8 (8q12.2), designated intractable disease |
| FZD5 | Syndromic coloboma + microcornea | Wnt signaling pathway receptor2) |
Systemic Complications
Section titled “Systemic Complications”Optic disc coloboma can be associated with the following systemic syndromes.
- CHARGE syndrome: A multiple organ malformation syndrome named after the initials of coloboma (C), heart defects (H), choanal atresia (A), growth retardation (R), genital hypoplasia (G), and ear abnormalities (E). The causative gene is CHD7, and it is designated as an intractable disease.
- Aicardi syndrome: Characterized by agenesis of the corpus callosum, epilepsy, and intellectual disability. It is more common in girls. Colobomas often appear as multiple blank spaces (lacunae) in the choroid and retina.
- Renal coloboma syndrome: Caused by PAX2 gene mutations. It is associated with renal and urinary tract malformations. Long-term follow-up for renal dysfunction is necessary. A PAX2 c.76delG frameshift mutation has been identified in a family with focal segmental glomerulosclerosis (FSGS), and the phenotypic spectrum is broader than previously thought1).
Optic disc coloboma can be associated with systemic syndromes such as CHARGE syndrome (multiple organ malformations due to CHD7 mutation, designated intractable disease), Aicardi syndrome (agenesis of the corpus callosum, epilepsy, female predominance), and renal coloboma syndrome (PAX2 mutation, renal and urinary tract malformations). In bilateral cases or when systemic findings are present, pediatric consultation and genetic counseling are recommended.
4. Diagnosis and examination methods
Section titled “4. Diagnosis and examination methods”Diagnostic methods
Section titled “Diagnostic methods”Diagnosis is possible based on ophthalmoscopic findings alone. Key diagnostic features include a well-defined white depression predominantly in the inferior part of the optic disc and characteristic abnormal vascular course (many vessels originating from the rim or within the depression). Ultrasound, MRI, CT, and optical coherence tomography (OCT) are used for definitive diagnosis.
Although sporadic cases are common, a family history may be present, so careful inquiry about family history is necessary.
Head MRI/CT is needed to check for intracranial malformations (e.g., agenesis of the corpus callosum). Pediatric consultation should be performed to confirm the absence of systemic complications such as CHARGE syndrome or Aicardi syndrome.
Examinations
Section titled “Examinations”| Examination | Purpose |
|---|---|
| Fundus examination (dilated) | Evaluation of optic disc morphology, vascular course, and retinal detachment |
| OCT (Optical Coherence Tomography) | Detailed evaluation of optic disc and macular structure |
| Ultrasound (B-mode) | Detection of retinal detachment when fundus is poorly visible |
| Visual field test | Evaluation of visual field defect patterns (e.g., superior field defect) |
| Brain MRI | Search for CNS complications such as corpus callosum agenesis and encephalocele |
| Renal ultrasound | Screening for renal coloboma syndrome |
| Genetic testing | Search for mutations in PAX2, CHD7, etc. (in bilateral or syndromic cases) |
Differential diagnosis
Section titled “Differential diagnosis”| Differential diagnosis | Key differentiating features |
|---|---|
| Morning glory syndrome | Glial proliferation at the center of the optic disc, radial vessels. Coloboma has inferiorly predominant excavation without glial proliferation. |
| Peripapillary staphyloma | Posterior bulging of the sclera surrounding the optic disc. Coloboma is a defect of the optic disc itself. |
| Optic disc PFV/PHPV (persistent fetal vasculature/persistent hyperplastic primary vitreous) | Associated with vitreous strands and retinal folds. Fundus findings differ from coloboma. |
| Megalopapilla | Large optic disc but nearly normal morphology. No cupping or abnormal vessel course.5) |
| Optic nerve hypoplasia | Small optic disc (DM/DD ratio ≥3.2). Coloboma has enlarged and excavated disc. |
| Glaucomatous optic atrophy | Progressive cupping enlargement and elevated intraocular pressure. Coloboma is non-progressive with normal IOP. |
5. Standard treatment
Section titled “5. Standard treatment”Optic disc coloboma itself is a congenital structural abnormality with no curative treatment. Symptomatic therapy is the mainstay, depending on the presence and type of complications.
Observation
Section titled “Observation”Since it is a non-progressive congenital anomaly, regular fundus examination is continued if there are no complications such as serous retinal detachment. In childhood, dilated ophthalmoscopy every 6 months to 1 year is recommended.
Management of Serous Retinal Detachment
Section titled “Management of Serous Retinal Detachment”There is no established treatment for serous retinal detachment, and spontaneous resolution has been reported. Observation for several months may be considered. If no improvement is seen after observation, surgical intervention is considered.
It is hypothesized that vitreous fluid enters the subretinal space due to structural abnormalities in the excavated area, and the possibility of cerebrospinal fluid inflow through communication between the excavation and the subarachnoid space has also been suggested.
Management of Rhegmatogenous Retinal Detachment
Section titled “Management of Rhegmatogenous Retinal Detachment”For rhegmatogenous retinal detachment, vitrectomy and photocoagulation around the excavation are performed. Postoperative visual prognosis is not necessarily good.
Retinal reattachment using fibrin glue has been performed for coloboma-related retinal detachment, and a technique of applying fibrin glue around retinal tears at the coloboma margin to strengthen adhesion has been reported 3). Some cases have achieved improvement to a final visual acuity of 20/50.
Amblyopia Treatment
Section titled “Amblyopia Treatment”For cases with poor vision, especially unilateral, refractive correction and occlusion therapy (patching of the healthy eye) are performed. Early intervention in childhood is important. However, the effect of amblyopia treatment is limited in cases of vision loss due to structural abnormalities of the optic nerve itself.
Systemic management
Section titled “Systemic management”- Cases with CHARGE syndrome: Multidisciplinary management involving cardiac surgery, otorhinolaryngology, endocrinology, etc. is necessary.
- Renal coloboma syndrome: Long-term follow-up of renal function is performed.
- Genetic counseling: Recommended in bilateral or syndromic cases.
Serous retinal detachment may resolve spontaneously, so observation for several months is performed first. For rhegmatogenous retinal detachment, vitrectomy and photocoagulation around the depression are performed. In recent years, there have been reports of adhesion reinforcement using fibrin glue. However, the visual prognosis after surgery is not necessarily good.
6. Pathophysiology and detailed pathogenesis
Section titled “6. Pathophysiology and detailed pathogenesis”Closure process of the embryonic fissure
Section titled “Closure process of the embryonic fissure”The optic cup is formed from the neuroectoderm at 4 weeks of gestation. An embryonic fissure (optic fissure) forms on the ventral side of the optic cup, through which the hyaloid artery passes. This fissure is completed at 5 weeks, and closure begins at 6 weeks. Closure starts near the equator and proceeds anteriorly (toward the iris) and posteriorly (toward the optic nerve), completing at 7 weeks. Localized failure of posterior closure results in optic nerve coloboma.
Epithelial-mesenchymal transition (EMT) is involved in the closure process. Neuroretinal epithelial cells at the margins of the embryonic fissure degrade the basement membrane, acquire mesenchymal traits, and fuse. Disruption of this process leads to coloboma6).
Molecular mechanisms
Section titled “Molecular mechanisms”The PAX2 gene is involved in determining the ventral aspect of the eye and is thought to be involved in closure of the embryonic fissure. PAX2 mutations cause renal coloboma syndrome. A PAX2 c.76delG frameshift mutation has been identified in an FSGS family, and the phenotype of renal coloboma syndrome is broader than previously thought1).
The FZD5 gene encodes a receptor of the Wnt signaling pathway. Loss-of-function mutations impair ligand-dependent activation of Wnt signaling, leading to failure of embryonic fissure closure and microcornea. It follows a recessive inheritance pattern2).
The CHD7 gene encodes a chromatin remodeling factor and is involved in neural crest cell differentiation and migration. Mutations cause CHARGE syndrome.
Mechanism of serous retinal detachment
Section titled “Mechanism of serous retinal detachment”It is hypothesized that vitreous humor flows into the subretinal space due to structural abnormalities in the colobomatous area. The possibility of cerebrospinal fluid inflow through communication between the coloboma and the subarachnoid space has also been suggested, which is considered one of the reasons why treatment of serous retinal detachment is difficult.
Prognosis and Course
Section titled “Prognosis and Course”Optic disc coloboma is a non-progressive, fixed congenital anomaly. The presence of retinal detachment is the main factor that worsens visual prognosis. Even after vitrectomy for rhegmatogenous retinal detachment, visual prognosis is often poor.
7. Latest Research and Future Perspectives
Section titled “7. Latest Research and Future Perspectives”Expansion of the Phenotypic Spectrum of PAX2 Mutations
Section titled “Expansion of the Phenotypic Spectrum of PAX2 Mutations”Hu et al. (2024) reported that a PAX2 c.76delG frameshift mutation was identified in a family with focal segmental glomerulosclerosis (FSGS) 1). The phenotype of renal coloboma syndrome is broader than previously thought, and the importance of renal function screening in cases of optic disc coloboma has been reaffirmed. It suggests expanding the indications for renal function evaluation in patients with coloboma.
FZD5 and Wnt Signaling Abnormalities
Section titled “FZD5 and Wnt Signaling Abnormalities”Cortes-Gonzalez et al. (2024) reported that a homozygous missense mutation in FZD5 (p.M160V) causes syndromic ocular coloboma and microcornea 2). Functional analysis confirmed that ligand-dependent activation of Wnt signaling is impaired in a recessive inheritance pattern. The genetic diagnostic rate for coloboma is less than 30%, and identification of novel causative genes is expected to improve diagnosis.
New Surgical Techniques for Retinal Detachment
Section titled “New Surgical Techniques for Retinal Detachment”Jain et al. (2024) reported performing retinal reattachment surgery with fibrin glue for coloboma-associated retinal detachment, achieving improvement to a final visual acuity of 20/50 3). The technique of applying fibrin glue around retinal tears at the coloboma margin to enhance adhesion is attracting attention as an adjunctive option to conventional vitrectomy plus photocoagulation. Accumulation of case numbers and verification of long-term outcomes are future challenges.
8. References
Section titled “8. References”-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.