
视神经乳头缺损
一目了然的要点
Section titled “一目了然的要点”1. 什么是视盘缺损?
Section titled “1. 什么是视盘缺损?”视盘缺损是一种先天性异常,表现为视盘异常扩大和边界清晰的白色凹陷。它由眼杯裂(胚胎裂)闭合不全引起,该裂通常在胚胎第7周闭合。视网膜血管并非从一个点起始,而是从凹陷边缘或凹陷内的不同部位起始。
当眼杯裂闭合不全局限于后部(视神经侧)时,发生视盘缺损。如果闭合不全从前到后更广泛,则形成虹膜至脉络膜缺损的谱系。在整个缺损谱系中,视盘缺损对应于眼杯裂闭合不全的后端,是虹膜缺损(前端)连续谱系的一部分,但也存在局限于视盘的孤立型。
ICD-10编码为H47.319(视神经)。
与牵牛花综合征的鉴别很重要。牵牛花综合征在视盘中心可见胶质增生组织,血管呈放射状走行。视盘缺损可通过无胶质增生、凹陷以下方为主以及血管从凹陷边缘或凹陷内不同部位起始来鉴别。
2. 主要症状和临床所见
Section titled “2. 主要症状和临床所见”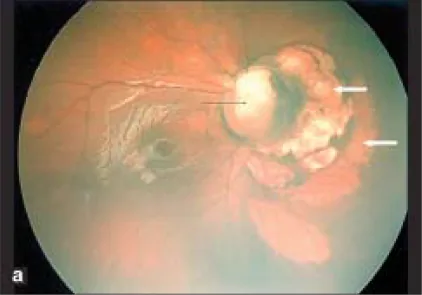
视力取决于乳头黄斑束是否被卷入缺损及其程度。从超过1.0到不良不等,但即使黄斑部无异常,由于视神经异常,视力下降的病例也很多。
- 视力下降:取决于乳头黄斑束的卷入程度。视力不良病例可能出现废用性斜视。
- 视野缺损:常对应于乳头下方的缺损,产生上方视野缺损。
- 斜视:视力不良病例可继发废用性斜视。
眼底可见以视神经乳头下方为中心的缺损,以及眼球下方的视网膜脉络膜缺损。伴有血管走行异常,由于视网膜中央动脉在乳头后方分支,可见多条视网膜动脉从乳头发出。乳头上方的边缘常残留,即使整体凹陷,典型情况下下方比上方更明显。
乳头区域凹陷,乳头缺失或部分缺损,周围的脉络膜、视网膜色素上皮(RPE)和巩膜也变薄。乳头区域凹陷的下方,存在由于胚胎裂闭合不全导致的视网膜脉络膜萎缩和纹理样改变。
视神经乳头缺损根据合并范围分类如下。
- 单纯视神经乳头缺损型:由于眼杯裂后部局限性闭合不全所致。
- 合并视网膜脉络膜缺损型:显示更广泛范围的眼杯裂闭合不全。
- 虹膜/睫状体缺损联合型:从前端到后端连续的广泛型。
- Fuchs缺损:轻微型,在视盘下方显示类似圆锥的萎缩性病变。视力通常相对保留。
- 常合并虹膜缺损和脉络膜缺损。
- 当合并脉络膜缺损且凹陷区域较大时,可能出现小眼球。
- 浆液性视网膜脱离:即使单独视盘缺损也可能发生。
- 孔源性视网膜脱离:在复杂的视网膜脉络膜缺损病例中可能继发。
- 经巩膜滤过导致的低眼压:已有巩膜缺损处房水渗漏的病例报道7)。
3. 病因、流行病学、风险因素
Section titled “3. 病因、流行病学、风险因素”患病率报道为3~8/100,000。单眼和双眼病例比例相当,多为散发病例,但常有家族史。已报道多种遗传方式,包括常染色体显性、常染色体隐性和X连锁遗传4)。
缺损的整体遗传诊断率仍低于30%5)。许多未发现基因突变的散发病例提示环境因素和多个修饰基因的参与。
胚胎裂(眼杯裂)在胚胎第4周神经外胚层形成眼杯时出现于腹侧。第5周完成,第6周开始闭合。闭合从赤道部附近向前方(虹膜侧)和后方(视神经侧)进行,第7周完成。后方局部闭合不全导致视盘缺损。
| 基因 | 相关疾病 | 备注 |
|---|---|---|
| PAX2 | 肾缺损综合征(renal coloboma syndrome) | 参与眼球腹侧决定和胚胎裂闭合1) |
| CHD7 | CHARGE综合征 | 第8号染色体(8q12.2),指定难治性疾病 |
| FZD5 | 综合征性缺损+小角膜 | Wnt信号通路受体2) |
视盘缺损可合并以下全身综合征。
- CHARGE综合征:以缺损(C)、心脏畸形(H)、后鼻孔闭锁(A)、生长迟缓(R)、生殖器发育不全(G)、耳部异常(E)的首字母命名的多器官畸形综合征。致病基因为CHD7,被认定为指定难治性疾病。
- Aicardi综合征:伴有胼胝体缺失、癫痫、精神发育迟滞。多见于女童。缺损常表现为脉络膜视网膜上的多个空白区(lacunae)。
- 肾缺损综合征:由PAX2基因突变引起。合并肾尿路发育异常。需要长期随访肾功能。PAX2的c.76delG移码突变已在局灶节段性肾小球硬化症(FSGS)家系中被鉴定,表型谱比以往认为的更广1)。
视盘缺损可能合并全身综合征,如CHARGE综合征(CHD7基因突变导致的多器官畸形,指定难治性疾病)、Aicardi综合征(胼胝体缺失、癫痫、女童为主)、肾缺损综合征(PAX2突变,肾尿路发育异常)。双眼性或存在全身表现时,建议儿科会诊和遗传咨询。
4. 诊断与检查方法
Section titled “4. 诊断与检查方法”仅凭检眼镜所见即可诊断。关键诊断要点为视盘下方为主的边界清晰的白色凹陷及特征性血管走行异常(从凹陷边缘或凹陷内发出大量血管)。确诊需使用超声、MRI、CT、光学相干断层扫描(OCT)。
虽然散发病例较多,但也可能存在家族史,因此需要仔细询问家族史。
需进行头部MRI/CT以检查是否合并颅内畸形(如胼胝体缺失)。应请儿科会诊以确认是否存在CHARGE综合征、Aicardi综合征等全身并发症。
| 检查 | 目的 |
|---|---|
| 眼底检查(散瞳) | 评估视盘形态、血管走行及视网膜脱离 |
| OCT(光学相干断层扫描) | 详细评估视盘和黄斑结构 |
| 超声检查(B超) | 眼底观察困难时检测视网膜脱离 |
| 视野检查 | 评估视野缺损模式(如上视野缺损) |
| 头部MRI | 检查中枢神经系统并发症,如胼胝体缺如和脑膨出 |
| 肾脏超声 | 筛查肾缺损综合征 |
| 基因检测 | 检测PAX2、CHD7等基因突变(双侧或综合征性病例) |
| 鉴别诊断 | 鉴别要点 |
|---|---|
| 牵牛花综合征(morning glory syndrome) | 视乳头中心有胶质增生组织,血管呈放射状走行。而缺损(coloboma)为下方为主的凹陷,无胶质增生。 |
| 视乳头周围葡萄肿(peripapillary staphyloma) | 围绕视乳头的巩膜向后膨隆。缺损(coloboma)是视乳头本身的缺损。 |
| 视乳头PFV/PHPV(原始玻璃体动脉残留) | 伴有玻璃体索条和视网膜皱襞。眼底表现与缺损(coloboma)不同。 |
| 大视乳头(megalopapilla) | 视乳头直径大,但形态接近正常。无凹陷或血管走行异常。5) |
| 视神经发育不全 | 视乳头小(DM/DD比≥3.2)。缺损(coloboma)为视乳头扩大和凹陷。 |
| 青光眼性视神经萎缩 | 进行性凹陷扩大和眼压升高。缺损(coloboma)为非进行性,眼压正常。 |
5. 标准治疗方法
Section titled “5. 标准治疗方法”视盘缺损本身是一种先天性结构异常,没有根治性治疗方法。主要根据并发症的有无和类型进行对症治疗。
由于是非进行性先天异常,如果没有浆液性视网膜脱离等并发症,则继续定期进行眼底检查。儿童期建议每半年至一年进行一次散瞳眼底检查。
浆液性视网膜脱离的处理
Section titled “浆液性视网膜脱离的处理”浆液性视网膜脱离的治疗尚无定论,有自然消退的病例报告。有时会观察数月。如果观察后无改善,则考虑手术干预。
推测是由于凹陷处的结构异常导致玻璃体液进入视网膜下腔,也有研究表明凹陷与蛛网膜下腔相通可能导致脑脊液流入。
孔源性视网膜脱离的处理
Section titled “孔源性视网膜脱离的处理”对于孔源性视网膜脱离,进行玻璃体手术和凹陷周围的光凝治疗。术后视力预后不一定良好。
有报道使用纤维蛋白胶进行视网膜复位术治疗缺损相关性视网膜脱离,在缺损边缘的视网膜裂孔周围涂抹纤维蛋白胶以加强粘附 3)。部分病例最终视力改善至20/50。
对于视力不良的病例,尤其是单眼性,进行屈光矫正和遮盖疗法(遮盖健眼)。儿童期早期干预很重要。但由于视神经本身结构异常导致的视力下降,弱视治疗效果有限。
浆液性视网膜脱离有时可自然消退,因此首先进行数月的观察。对于孔源性视网膜脱离,进行玻璃体手术和凹陷周围的光凝。近年来也有使用纤维蛋白胶加强黏附的报道。但术后视力预后不一定良好。
6. 病理生理学与详细发病机制
Section titled “6. 病理生理学与详细发病机制”胚胎裂的闭合过程
Section titled “胚胎裂的闭合过程”眼杯在胚胎第4周由神经外胚层形成。眼杯腹侧出现胚胎裂(眼杯裂),玻璃体动脉通过其中。此裂在第5周完成,第6周开始闭合。闭合从赤道部附近开始,向前方(虹膜侧)和后方(视神经侧)推进,第7周完成。后方局部闭合不全导致视神经缺损。
闭合过程涉及上皮-间充质转化(EMT)。胚胎裂边缘的神经视网膜上皮细胞降解基底膜,获得间充质特性并融合。此过程障碍导致缺损6)。
PAX2基因参与眼球腹侧的确定,并被认为参与胚胎裂的闭合。PAX2突变导致肾缺损综合征。在FSGS家系中发现了PAX2 c.76delG移码突变,肾缺损综合征的表型比以前认为的更广泛1)。
FZD5基因编码Wnt信号通路的受体。功能丧失型突变损害Wnt信号的配体依赖性激活,导致胚胎裂闭合不全和小角膜。呈隐性遗传模式2)。
CHD7基因编码染色质重塑因子,参与神经嵴细胞的分化和迁移。突变导致CHARGE综合征。
浆液性视网膜脱离的机制
Section titled “浆液性视网膜脱离的机制”推测由于凹陷部位的结构异常,玻璃体液流入视网膜下腔。也有提示通过凹陷与蛛网膜下腔的交通导致脑脊液流入的可能性,这被认为是浆液性视网膜脱离治疗困难的原因之一。
视神经乳头缺损是一种非进行性的固定先天异常。合并视网膜脱离是恶化视力预后的主要因素。即使对孔源性视网膜脱离进行玻璃体手术后,视力预后也往往不佳。
7. 最新研究与未来展望
Section titled “7. 最新研究与未来展望”PAX2突变表型谱的扩展
Section titled “PAX2突变表型谱的扩展”Hu等人(2024)报告在一个局灶节段性肾小球硬化症(FSGS)家系中鉴定出PAX2的c.76delG移码突变1)。肾缺损综合征的表型比以前认为的更广泛,视神经乳头缺损病例中肾功能筛查的重要性再次得到证实。提示应扩大缺损患者肾功能评估的适应症。
FZD5与Wnt信号传导异常
Section titled “FZD5与Wnt信号传导异常”Cortes-Gonzalez等人(2024)报告FZD5的纯合错义突变(p.M160V)导致综合征性眼缺损和小角膜2)。功能分析证实,隐性遗传模式下Wnt信号的配体依赖性激活受损。缺损的遗传学诊断率低于30%,新致病基因的鉴定有望提高诊断水平。
视网膜脱离的新手术技术
Section titled “视网膜脱离的新手术技术”Jain等人(2024)报告对缺损相关性视网膜脱离施行纤维蛋白胶联合视网膜复位术,最终视力改善至20/503)。在缺损边缘的视网膜裂孔周围涂抹纤维蛋白胶以增强粘附的技术,作为传统玻璃体手术加光凝的辅助选择受到关注。病例数的积累和长期结果的验证是未来的课题。
8. 参考文献
Section titled “8. 参考文献”-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.