Aicardi syndrome is a congenital rare disease first described in 1967 by French neurologist Jean Aicardi. It is presumed to be X-linked dominant, and almost all patients are girls. In boys, it is lethal in the hemizygous state, so only a few cases have been reported in boys with XXY karyotype (Klinefelter syndrome).
The incidence is estimated at about 1:110,000 births, and the number of affected individuals worldwide is approximately 4,000 1). All cases are de novo mutations, with no parent-to-child transmission, and the recurrence risk for siblings is less than 1% 1).
The classic triad consists of the following three features 1):
Infantile spasms: onset around 3–4 months of age.
Chorioretinal lacunae: Bilateral round fundus lesions. A finding specific to this disease.
Agenesis of the corpus callosum: Partial or complete absence.
The prognosis is poor. The average survival age is 18 years, and the probability of surviving to age 27 is reported to be 0.62% 1).
QDoes Aicardi syndrome occur in boys?
A
This disease occurs almost exclusively in girls. It is presumed to be X-linked dominant, as it is lethal in hemizygous males. However, there are a few reported cases worldwide in boys with XXY karyotype (Klinefelter syndrome).
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Retcam photo of right eye shows optic disc coloboma (black arrow) and dome shaped loci of pale areas with sharp borders nasal to the optic disc suggestive of chorio retinal lacunae (white arrows).
The initial symptom of this disease is usually infantile spasms appearing around 3-4 months of age. Epilepsy often becomes drug-resistant and is accompanied by various seizure types.
Epileptic seizures: Initially presenting as infantile spasms, progressing to drug resistance. In one case, generalized convulsions occurred 3-4 times a day (each lasting 20-25 minutes) at 4 months of age 1). In another case, frequent eyelid myoclonia seizures were observed from 1 month of age 2).
Psychomotor developmental delay: Accompanied by severe intellectual disability, often making independent movement and language acquisition difficult.
Gastrointestinal dysfunction: Gastrointestinal symptoms such as constipation are present in over 90% of cases 1).
Visual impairment: Visual impairment due to fundus lesions, corpus callosum agenesis, and cortical malformations.
Case reports have documented an example with preretinal hemorrhage and 360-degree peripheral avascular zone in the right eye, and stalk tissue (fibrovascular stalk) with tractional retinal detachment in the left eye2). It can also cause cortical visual impairment (CVI)3).
Agenesis of the corpus callosum: Partial or complete absence is present in all cases1). Thinning of the corpus callosum (dysgenesis) has also been reported as a variant2).
Cerebral cortical malformations: Polymicrogyria (Barkovich type 2), periventricular gray matter heterotopia, and multicystic lesions are confirmed on MRI1).
EEG findings: Characteristic pattern of high-voltage polymorphic rhythms with multifocal spikes and wave discharges1).
Skeletal abnormalities: Thoracic vertebral fusion (T9–T10) and butterfly vertebra (T8) are seen in 40–60% of cases1).
QDo chorioretinal lacunae change over time?
A
They may progress. New lacunae have been reported to become visible after ophthalmic surgical intervention, and their size or number may increase over time2). Regular fundus examination follow-up is important.
Aicardi syndrome is presumed to be X-linked dominant, but the causative gene has not been identified to date1). All cases are de novo mutations, and familial occurrence is essentially absent. The recurrence risk for siblings is less than 1%, and genetic counseling is recommended when planning future children1).
In a recent case report, a TREX1 gene mutation (c.292_293insA, p.(Cys99Metfs)) was detected in one patient2). TREX1 is located on chromosome 3, which partially contradicts the X-linked hypothesis, so the location of the causative gene remains under debate.
Inheritance pattern: X-linked dominant (presumed). Hemizygous males are lethal in utero.
Nature of mutation: All de novo mutations. No inheritance (parent-to-child transmission).
Since the causative gene has not been identified, the diagnosis of Aicardi syndrome is primarily clinical. Confirmation of the classic triad is central to diagnosis1).
Infantile spasms
Chorioretinal lacunae
Agenesis of the corpus callosum
Even if only two of the three cardinal features are present, diagnosis is possible using the expanded diagnostic criteria established in 1999.
The components of the expanded diagnostic criteria are shown below.
Multidisciplinary collaboration: Neurology, ophthalmology, orthopedics, and genetics departments need to collaborate for diagnosis 1).
QCan genetic testing provide a definitive diagnosis?
A
Currently, no causative gene has been identified that allows definitive diagnosis, so genetic testing alone cannot provide a definitive diagnosis 1). Clinical diagnosis based on a combination of clinical symptoms, fundus findings, and imaging findings is the main approach. Advances in comprehensive genomic analysis are expected to identify causative genes in the future.
There is no curative treatment. The goals of treatment consist of three pillars: seizure control, management of ophthalmic complications, and developmental support through rehabilitation.
Seizure management
First-line drugs: Phenytoin, levetiracetam, clobazam, etc. are used 1).
Refractory cases: Cannabidiol (CBD), ketogenic diet therapy, corpus callosotomy, and vagus nerve stimulation are attempted 1).
Ophthalmic intervention
Laser photocoagulation: Performed on peripheral avascular retina to prevent progression of proliferative retinopathy 2).
Reported cases of surgical interventions for ophthalmic complications are presented.
Eye
Findings
Treatment
Right eye
Preretinal hemorrhage, avascular zone 360 degrees
Laser photocoagulation
Left eye
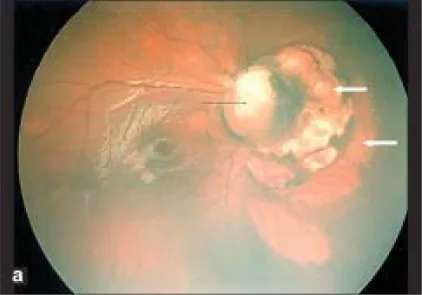
Stalk tissue, TRD
23G vitrectomy
After vitrectomy, recovery of axial length growth was confirmed; in the left eye, axial length grew from 17.45 mm at 1 month of age to 24.41 mm at 26 months of age 2). In addition, there are reports of cases where chorioretinal lacunae became newly visible after surgery, contributing to a definitive diagnosis 2).
QIs ophthalmic surgical treatment possible?
A
Yes, it is possible. Laser photocoagulation for peripheral avascular retina and 23-gauge vitrectomy for tractional retinal detachment may be effective 2). There are reports of accelerated ocular growth after surgery. However, the number of cases is small, and careful management at a specialized facility is required.
De novo mutations on the X chromosome are presumed to be the cause of this disease. The pattern of X-chromosome inactivation (lyonization) is thought to produce phenotypic variability even with the same mutation. In males, hemizygosity leads to lethality in the embryonic period, and only males with an XXY karyotype can survive.
Recently, a TREX1 gene mutation (c.292_293insA, p.(Cys99Metfs)) was detected in one case diagnosed with this disease 2). Since TREX1 is located on chromosome 3, this may contradict the X-linked hypothesis, suggesting the possibility of non-X-linked causative genes.
Histologically, chorioretinal lacunae are characterized by defects in the retinal pigment epithelium (RPE) extending from the choriocapillaris to the bare sclera2). The defect area lacks RPE and choriocapillaris, and dysplastic undifferentiated neuroretina is observed.
Persistence of the fetal vasculature is thought to contribute to the formation of fibrovascular stalk tissue and peripheral avascular retina2). These abnormal vessels cause tractional retinal detachment.
Cortical visual impairment (CVI) arises as an acquired visual function decline due to brain structural abnormalities such as polymicrogyria, heterotopic gray matter, and agenesis of the corpus callosum 3).
Brain malformations (polymicrogyria, agenesis of the corpus callosum) are based on impaired neuronal migration during the embryonic period. The molecular mechanisms underlying this migration disorder remain unknown, and future research is awaited along with identification of causative genes.
7. Latest Research and Future Prospects (Investigational Reports)
The causative gene remains unidentified, but detection of TREX1 gene mutations 2) is a new clue suggesting the involvement of non-X-linked mutations. The widespread use of comprehensive genomic analysis (WES/WGS) is expected to accelerate the identification of causative genes. Verification of the X-linked and non-X-linked hypotheses is a major future challenge.
Recognition of Variant (Atypical) Aicardi Syndrome
Reports of variant cases showing only thinning (dysgenesis) rather than complete agenesis of the corpus callosum are accumulating 2). Even in cases that do not meet all three cardinal features, detailed fundus examination and application of expanded diagnostic criteria may improve diagnostic accuracy.
Kang et al. (2022) reported that laser photocoagulation and 23-gauge vitrectomy performed for bilateral vitreoretinopathy in Aicardi syndrome contributed to maintaining ocular growth and protecting visual function 2). They also recorded that chorioretinal lacunae became newly visible postoperatively, indicating that ophthalmic surgery can also aid in confirming the diagnosis.
There is growing recognition that early fundus screening and prompt intervention are important for protecting retinal function 2). Laser treatment to peripheral avascular areas may prevent the progression of proliferative retinopathy, and further case accumulation is desired.
Research on the application of cannabidiol (CBD) and ketogenic diet therapy for seizure control is advancing 1). Establishment of new treatment options for intractable epilepsy is anticipated.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.