กลุ่มอาการไอคาร์ดีเป็นโรคหายากแต่กำเนิดที่ถ่ายทอดทางพันธุกรรมแบบเด่นบนโครโมโซม X เกิดขึ้นเกือบเฉพาะในเด็กผู้หญิง โดยประมาณการอุบัติการณ์ประมาณ 1:110,000 การเกิด
สามลักษณะคลาสสิกคือ “อาการชักกระตุกในทารก, รอยหลุมคอรอยด์ และจอประสาทตา , การไม่มีคอร์ปัส คัลโลซัม” ซึ่งทั้งสามนี้เป็นแกนหลักของการวินิจฉัย
รอยหลุมคอรอยด์ และจอประสาทตา (รอยโรคที่จอตารูปกลมสีขาวเหลือง) เป็นลักษณะเฉพาะของโรคเกือบทั้งหมด พบได้ใน 70-90% ของผู้ป่วย
โรคลมชักมักพัฒนาไปสู่การดื้อยา ร่วมกับพัฒนาการทางจิตใจและการเคลื่อนไหวล่าช้า
ภาวะแทรกซ้อนทางตา (บริเวณจอประสาทตา ไร้หลอดเลือด, จอประสาทตาลอก แบบดึงรั้ง) อาจรักษาได้ด้วยเลเซอร์หรือการผ่าตัดน้ำวุ้นตา
ยังไม่มีการระบุยีนก่อโรคที่แน่ชัด การวินิจฉัยอาศัยอาการทางคลินิกเป็นหลัก
อายุขัยเฉลี่ยประมาณ 18 ปี การพยากรณ์โรคไม่ดี แต่มีความแตกต่างกันมากในแต่ละบุคคล
กลุ่มอาการไอคาร์ดี (Aicardi syndrome) เป็นโรคหายากแต่กำเนิดที่ได้รับการบรรยายครั้งแรกในปี ค.ศ. 1967 โดยนักประสาทวิทยาชาวฝรั่งเศส Jean Aicardi สันนิษฐานว่ามีการถ่ายทอดทางพันธุกรรมแบบเด่นบนโครโมโซม X และผู้ป่วยเกือบทั้งหมดเป็นเพศหญิง ในเพศชาย โรคนี้ทำให้เสียชีวิตในภาวะเฮมิไซกัส ดังนั้นจึงมีรายงานเพียงไม่กี่รายในเพศชายที่มีคาริโอไทป์ XXY (กลุ่มอาการไคลน์เฟลเตอร์)
อุบัติการณ์ประมาณ 1:110,000 การเกิด และจำนวนผู้ป่วยทั่วโลกประมาณ 4,000 คน 1) ทุกกรณีเกิดจากการกลายพันธุ์ใหม่ (de novo) ไม่มีการถ่ายทอดจากพ่อแม่สู่ลูก และความเสี่ยงที่จะเกิดซ้ำในพี่น้องน้อยกว่า 1% 1)
สามลักษณะคลาสสิกที่ทราบกันดีมีดังนี้ 1) :
อาการชักกระตุกในทารก (infantile spasms) : เริ่มมีอาการเมื่ออายุประมาณ 3-4 เดือนรอยโรคคอริโอเรตินาลาคูเน (chorioretinal lacunae) : รอยโรคที่จอตาทรงกลมทั้งสองข้าง เป็นลักษณะเฉพาะของโรคนี้ภาวะไม่มีคอร์ปัส คาโลซัม (agenesis of the corpus callosum) : การขาดหายไปบางส่วนหรือทั้งหมด
การพยากรณ์โรคไม่ดี อายุขัยเฉลี่ยคือ 18 ปี และความน่าจะเป็นที่จะมีชีวิตอยู่ถึง 27 ปีรายงานไว้ที่ 0.62% 1)
Q
กลุ่มอาการไอคาร์ดีเกิดขึ้นในเด็กผู้ชายด้วยหรือไม่?
A
โรคนี้เกิดขึ้นเกือบเฉพาะในเด็กผู้หญิงเท่านั้น สันนิษฐานว่าเป็นพันธุกรรมแบบเด่นบนโครโมโซม X เนื่องจากในเด็กผู้ชายที่เป็นเฮมิไซกัสจะทำให้เสียชีวิต อย่างไรก็ตาม มีรายงานผู้ป่วยในเด็กผู้ชายที่มีคาริโอไทป์ XXY (กลุ่มอาการไคลน์เฟลเตอร์) เพียงไม่กี่รายทั่วโลก
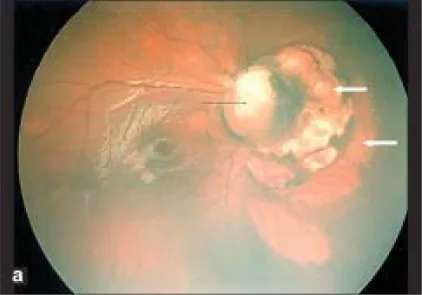
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PM
CI D: PMC2683450. License: CC BY.
ภาพ Retcam ของตาขวาแสดงคอโลโบมาของจานประสาทตา (ลูกศรดำ) และบริเวณสีซีดรูปโดมที่มีขอบเขตชัดเจนทางด้านจมูกของจานประสาทตา ซึ่งบ่งชี้ถึงรอยโรคคอริโอเรตินาลาคูเน (ลูกศรขาว)
อาการเริ่มแรกของโรคนี้มักเป็นอาการชักแบบเกร็งกระตุกในทารก (infantile spasms) ซึ่งปรากฏเมื่ออายุประมาณ 3-4 เดือน อาการชักมักกลายเป็นดื้อต่อยาและมีรูปแบบการชักที่หลากหลายร่วมด้วย
อาการชักจากโรคลมชัก : เริ่มต้นด้วยอาการชักแบบเกร็งกระตุกในทารกและพัฒนาไปสู่การดื้อยา ในรายงานผู้ป่วยรายหนึ่ง อาการชักเกร็งกระตุกทั้งตัวเกิดขึ้น 3-4 ครั้งต่อวัน (แต่ละครั้งนาน 20-25 นาที) เมื่ออายุ 4 เดือน 1) ในอีกรายงานหนึ่ง พบอาการชักแบบกระพริบตาบ่อยครั้งตั้งแต่อายุ 1 เดือน 2) พัฒนาการทางจิตใจและการเคลื่อนไหวล่าช้า : ร่วมกับความบกพร่องทางสติปัญญาระดับรุนแรง มักยากที่จะบรรลุการเคลื่อนไหวอย่างอิสระหรือการได้มาซึ่งภาษาความผิดปกติของระบบทางเดินอาหาร : อาการทางเดินอาหารเช่นท้องผูกพบได้มากกว่า 90% ของผู้ป่วย 1) ความบกพร่องทางการมองเห็น : ความบกพร่องทางการมองเห็น เกิดจากรอยโรคที่จอตา ภาวะไม่มีคอร์ปัส คาโลซัม และความผิดปกติของคอร์เทกซ์
ในบรรดาอาการทางจักษุวิทยาของโรคนี้ รอยเว้าจอประสาทตา คอรอยด์ (choroidal retinal lacunae) ถือเป็นอาการเฉพาะโรค (pathognomonic)
รอยเว้าจอประสาทตาคอรอยด์
การกระจาย : เป็นทั้งสองข้าง กระจุกตัวรอบขั้วประสาทตาและขั้วหลัง แต่ยังขยายไปถึงบริเวณรอบนอก
ลักษณะ : รอยโรคสีขาวเหลืองถึงชมพู รูปกลมถึงรี ความชุก 70-90%1)
เนื้อเยื่อวิทยา : การสูญเสียเยื่อบุผิวรงควัตถุจอประสาทตา (RPE ) ซึ่งขยายจากชั้นคอรอยด์ ไปถึงชั้นตาขาว เปลือย2)
การดำเนินโรค : ขนาดและจำนวนอาจเพิ่มขึ้นตามเวลาหลังการผ่าตัด2)
อาการทางตาอื่นๆ
คอลโลโบมาของเส้นประสาทตา : พบร่วมประมาณ 44%
ตาเล็ก (microphthalmia) : พบประมาณ 20%
จอประสาทตา ไร้หลอดเลือดส่วนปลาย2)
จอประสาทตาลอก แบบดึงรั้ง (TRD)2)
ในรายงานผู้ป่วย มีการบันทึกกรณีที่มีเลือดออกก่อนจอประสาทตา และบริเวณไร้หลอดเลือดส่วนปลาย 360 องศาในตาขวา และเนื้อเยื่อก้าน (stalk tissue) ร่วมกับจอประสาทตาลอก แบบดึงรั้งในตาซ้าย2) นอกจากนี้ยังอาจทำให้เกิดความบกพร่องทางการมองเห็นจากสมอง ส่วนคอร์เทกซ์ (cortical visual impairment; CVI )3)
ภาวะไม่มีคอร์ปัส คาโลซัม (agenesis of corpus callosum) : การขาดหายบางส่วนหรือทั้งหมดพบในทุกราย1) นอกจากนี้ยังมีรายงานภาวะคอร์ปัส คาโลซัมเจริญผิดปกติ (dysgenesis) เป็นรูปแบบหนึ่ง2) ความผิดปกติของเปลือกสมอง : พบภาวะพอลิไมโครไจเรีย (polymicrogyria, ชนิดที่ 2 ตามการจำแนกของ Barkovich), ก้อนเนื้อเทาบริเวณรอบโพรงสมอง และถุงน้ำหลายช่องในการตรวจ MRI 1) ผลการตรวจ EEG : แสดงรูปแบบลักษณะเฉพาะของจังหวะหลายรูปแบบศักย์ไฟฟ้าสูงร่วมกับคลื่นแหลมหลายตำแหน่งและการปล่อยคลื่น 1) ความผิดปกติของโครงกระดูก : พบใน 40-60% ของผู้ป่วย ได้แก่ การเชื่อมติดของกระดูกสันหลังส่วนอก (T9-T10), กระดูกสันหลังรูปผีเสื้อ (T8) และอื่นๆ 1)
Q
รอยโรคคอริโอเรตินอลลาคูนาเปลี่ยนแปลงตามเวลาหรือไม่?
A
อาจมีการดำเนินโรค มีรายงานว่ารอยโรคใหม่อาจปรากฏขึ้นหลังการผ่าตัดตา หรือขนาดและจำนวนเพิ่มขึ้นตามเวลา 2) การติดตามผลด้วยการตรวจอวัยวะรับภาพเป็นประจำเป็นสิ่งสำคัญ
กลุ่มอาการไอคาร์ดีสันนิษฐานว่าเป็นโรคถ่ายทอดทางพันธุกรรมแบบเด่นบนโครโมโซม X แต่ยังไม่มีการระบุยีนก่อโรคในปัจจุบัน 1) ทุกกรณีเป็นการกลายพันธุ์ใหม่ (de novo) และโดยทั่วไปไม่พบการเกิดในครอบครัว ความเสี่ยงของการเกิดซ้ำในพี่น้องน้อยกว่า 1% และแนะนำให้วางแผนการมีบุตรคนต่อไปหลังจากปรึกษาทางพันธุกรรม 1)
ในรายงานผู้ป่วยล่าสุด มีการตรวจพบการกลายพันธุ์ของยีน TREX1 (c.292_293insA, p.(Cys99Metfs)) ในหนึ่งราย 2) TREX1 อยู่บนโครโมโซมคู่ที่ 3 ซึ่งขัดแย้งบางส่วนกับสมมติฐานการเชื่อมโยงกับโครโมโซม X ดังนั้นจึงยังคงมีการถกเถียงเกี่ยวกับตำแหน่งของยีนก่อโรค
รูปแบบการถ่ายทอด : เด่นบนโครโมโซม X (สันนิษฐาน) เพศชายที่มีเฮมิไซกัสจะเสียชีวิตในระยะทารกในครรภ์ลักษณะการกลายพันธุ์ : ทั้งหมดเป็นการกลายพันธุ์ใหม่ (de novo) ไม่มีการถ่ายทอดทางพันธุกรรม (จากพ่อแม่สู่ลูก)ความเสี่ยงของการเกิดซ้ำ : น้อยกว่า 1% ในพี่น้อง 1)
เนื่องจากยังไม่มีการระบุยีนก่อโรค การวินิจฉัยกลุ่มอาการไอคาร์ดีจึงขึ้นอยู่กับ การวินิจฉัยทางคลินิก เป็นหลัก การยืนยันสามลักษณะคลาสสิกต่อไปนี้เป็นหัวใจสำคัญของการวินิจฉัย 1)
อาการชักกระตุกในทารก (infantile spasms)
รอยเว้าคอรอยด์ และจอประสาทตา (chorioretinal lacunae)
ภาวะไม่มีคอร์ปัส คัลโลซัม (agenesis of the corpus callosum)
แม้ว่าจะมีเพียงสองในสามอาการหลัก ก็สามารถวินิจฉัยได้โดยใช้เกณฑ์การวินิจฉัยแบบขยายที่กำหนดขึ้นในปี 1999
องค์ประกอบของเกณฑ์การวินิจฉัยแบบขยายมีดังนี้
การจำแนก รายการหลัก ลักษณะสำคัญ คอโลโบมาของเส้นประสาทตา , ความผิดปกติของคอร์เทกซ์, สสารสีเทานอกตำแหน่ง, ถุงน้ำในกะโหลกศีรษะ, ติ่งเนื้อของคอรอยด์ เพล็กซัส ลักษณะสนับสนุน ความผิดปกติของกระดูกสันหลังและซี่โครง, ตาเล็ก, EEG แยกซีก, ความไม่สมมาตรของสมองซีก, ความผิดปกติของหลอดเลือด
สามารถยืนยันการวินิจฉัยได้ด้วยสองในสามอาการหลัก + ลักษณะสำคัญหรือสนับสนุนอย่างน้อยสองข้อ
MRI (ศีรษะ) : ยืนยันภาวะไม่มีคอร์ปัส คัลโลซัม ประเมินภาวะพอลิไมโครไจเรีย, สสารสีเทานอกตำแหน่ง, การขยายของโพรงสมองข้าง, ถุงน้ำทาลามัสทั้งสองข้าง, ภาวะฮิปโปแคมปัสเจริญไม่เต็มที่ ฯลฯ1) 2) EEG (คลื่นไฟฟ้าสมอง) : ยืนยันรูปแบบจังหวะหลายรูปแบบแรงดันสูงและคลื่นและสไปก์หลายจุด1) การตรวจอวัยวะรับภาพ (Fundus examination) : ยืนยันการมีอยู่ของ lacuna จอประสาทตา -คอรอยด์ การตรวจหลอดเลือดด้วยฟลูออเรสซีน (FA ) มีประโยชน์ในการประเมินบริเวณที่ไม่มีหลอดเลือด 2) เอกซเรย์กระดูกสันหลังส่วนอก (AP) : ยืนยันความผิดปกติของโครงกระดูก (กระดูกสันหลังเชื่อมติดกัน, กระดูกสันหลังรูปผีเสื้อ) 1) การทำงานร่วมกันแบบสหสาขา : แพทย์ระบบประสาท จักษุแพทย์ ศัลยกรรมกระดูก และแพทย์พันธุศาสตร์ต้องร่วมมือกันในการวินิจฉัย 1)
Q
การตรวจทางพันธุกรรมสามารถยืนยันการวินิจฉัยได้หรือไม่?
A
ในปัจจุบัน ยังไม่มีการระบุยีนก่อโรคที่สามารถยืนยันการวินิจฉัยได้ ดังนั้นจึงไม่สามารถวินิจฉัยได้จากการตรวจทางพันธุกรรมเพียงอย่างเดียว 1) การวินิจฉัยทางคลินิกโดยอาศัยอาการทางคลินิก ผลการตรวจอวัยวะรับภาพ และผลการตรวจภาพทางรังสีร่วมกันเป็นหลัก ด้วยความก้าวหน้าของการวิเคราะห์จีโนมแบบครอบคลุม คาดว่าจะสามารถระบุยีนก่อโรคได้ในอนาคต
ไม่มีการรักษาที่หายขาด เป้าหมายของการรักษาประกอบด้วยสามเสาหลัก: การควบคุมอาการชัก การจัดการภาวะแทรกซ้อนทางจักษุวิทยา และการสนับสนุนพัฒนาการผ่านการฟื้นฟูสมรรถภาพ
การจัดการโรคลมชัก
ยาทางเลือกแรก : ฟีนิโทอิน, เลเวทิราเซแทม, โคลบาแซม และอื่นๆ 1)
กรณีดื้อยา : อาจลองใช้แคนนาบิไดออล (CBD), อาหารคีโตเจนิก, การผ่าตัดตัดคอร์ปัส คัลโลซัม, การกระตุ้นเส้นประสาทเวกัส 1)
การแทรกแซงทางจักษุวิทยา
การจี้ด้วยเลเซอร์ : ทำบนจอประสาทตา ส่วนปลายที่ไม่มีหลอดเลือดเพื่อป้องกันการลุกลามของจอประสาทตา ผิดปกติชนิดมีเส้นเลือดงอก 2)
การตัดน้ำวุ้นตา : การตัดน้ำวุ้นตา 23G ทำสำหรับจอประสาทตาลอก ชนิดดึงรั้ง (TRD) 2)
การฟื้นฟูสมรรถภาพ
เริ่มต้นเร็ว : เริ่มกายภาพบำบัด กิจกรรมบำบัด การบำบัดการพูด และการบำบัดทางสายตาทันทีหลังการวินิจฉัย 1)
การดูแลผู้มีสายตาเลือนราง
ต่อไปนี้เป็นตัวอย่างการผ่าตัดแทรกแซงสำหรับภาวะแทรกซ้อนทางจักษุวิทยา
ตา ผลการตรวจ การรักษา ตาขวา เลือดออกก่อนจอประสาทตา และบริเวณไร้หลอดเลือด 360 องศา การจี้แข็งด้วยเลเซอร์ ตาซ้าย เนื้อเยื่อก้านและจอประสาทตาลอก แบบดึงรั้ง การตัดน้ำวุ้นตา 23G
หลังการตัดน้ำวุ้นตา พบว่าการเจริญเติบโตของความยาวแกนตา ฟื้นตัวได้ ในตาซ้าย ความยาวแกนตา เพิ่มขึ้นจาก 17.45 มม. เมื่ออายุ 1 เดือน เป็น 24.41 มม. เมื่ออายุ 26 เดือน2) นอกจากนี้ยังมีรายงานกรณีที่พบรอยเว้าคอรอยด์ และจอประสาทตา ใหม่หลังการผ่าตัด ซึ่งช่วยยืนยันการวินิจฉัย2)
โรคลมชักมักดื้อต่อยาหลายชนิด จึงจำเป็นต้องมีการจัดการแบบสหสาขาวิชาชีพในระยะยาว
รายงานการผ่าตัดทางตาในกลุ่มอาการไอคาร์ดีมีจำกัด จำเป็นต้องพิจารณาอย่างรอบคอบตามประสบการณ์ของสถานพยาบาล
หลังผ่าตัด ควรสังเกตอวัยวะตาอย่างสม่ำเสมอเพื่อติดตามความคืบหน้าของลาคูนาและการปรากฏของเส้นเลือดใหม่
Q
การรักษาด้วยการผ่าตัดทางตาเป็นไปได้หรือไม่?
A
เป็นไปได้ การจี้ด้วยเลเซอร์ที่จอประสาทตา ไร้หลอดเลือดส่วนปลาย และการตัดแก้วตา 23G สำหรับจอประสาทตาลอก แบบดึงรั้งอาจมีประสิทธิภาพในบางกรณี2) มีรายงานการกระตุ้นการเจริญเติบโตของลูกตาหลังผ่าตัด อย่างไรก็ตาม จำนวนผู้ป่วยมีน้อย จำเป็นต้องจัดการอย่างระมัดระวังในสถานพยาบาลเฉพาะทาง
การกลายพันธุ์แบบเดอโนโวบนโครโมโซม X สันนิษฐานว่าเป็นสาเหตุของโรคนี้ รูปแบบการหยุดการทำงานของโครโมโซม X (ไลโอไนเซชัน) เชื่อว่าทำให้เกิดความหลากหลายของฟีโนไทป์แม้จะมีการกลายพันธุ์เดียวกัน ในเพศชาย เนื่องจากเป็นเฮมิไซกัส ทำให้เสียชีวิตในระยะตัวอ่อน และมีเพียงเพศชายที่มีคาริโอไทป์ XXY เท่านั้นที่สามารถรอดชีวิตได้
เมื่อเร็วๆ นี้ มีการตรวจพบการกลายพันธุ์ของยีน TREX1 (c.292_293insA, p.(Cys99Metfs)) ในผู้ป่วย 1 รายที่ได้รับการวินิจฉัยว่าเป็นโรคนี้2) เนื่องจาก TREX1 อยู่บนโครโมโซม 3 สิ่งนี้อาจขัดแย้งกับสมมติฐานที่เชื่อมโยงกับ X ซึ่งบ่งชี้ถึงความเป็นไปได้ของยีนก่อโรคที่ไม่เชื่อมโยงกับ X
ในฐานะลักษณะทางเนื้อเยื่อวิทยาของลาคูนาคอรอยด์ -จอประสาทตา ได้รับการยืนยันว่าข้อบกพร่องของเยื่อบุผิวรงควัตถุจอประสาทตา (RPE ) ขยายจากแผ่นเส้นเลือดฝอยคอรอยด์ ไปยังชั้นตาขาว เปลือย2) ไม่มีเยื่อบุผิวรงควัตถุจอประสาทตา และแผ่นเส้นเลือดฝอยคอรอยด์ ในตำแหน่งที่บกพร่อง และพบความผิดปกติของจอประสาทตา ประสาทที่ยังไม่แยกความแตกต่าง
ส่วนที่เหลือของระบบหลอดเลือดของทารกในครรภ์ที่คงอยู่ (persistent fetal vasculature) เชื่อว่ามีส่วนเกี่ยวข้องในการก่อตัวของก้านเนื้อเยื่อเส้นใยหลอดเลือด (stalk tissue) และจอประสาทตา ไร้หลอดเลือดส่วนปลาย2) หลอดเลือดที่ผิดปกติเหล่านี้ทำให้เกิดจอประสาทตาลอก แบบดึงรั้ง
ความบกพร่องทางการมองเห็น จากเปลือกสมอง (CVI ) คือการลดลงของการทำงานทางการมองเห็น ที่เกิดขึ้นภายหลังเนื่องจากความผิดปกติของโครงสร้างสมอง เช่น พอลิไมโครไจเรีย (polymicrogyria) สสารสีเทาต่างที่ (heterotopic gray matter) และภาวะไม่มีคอร์ปัส คัลโลซัม (agenesis of corpus callosum)3)
ความผิดปกติของสมอง (พอลิไมโครไจเรีย, ภาวะไม่มีคอร์ปัส คัลโลซัม) มีพื้นฐานมาจากความผิดปกติของการย้ายที่ของเซลล์ประสาทในระยะตัวอ่อน กลไกระดับโมเลกุลที่ทำให้เกิดความผิดปกติของการย้ายที่นี้ยังไม่ทราบแน่ชัด และรอการวิจัยในอนาคตพร้อมกับการระบุยีนก่อโรค
ยีนก่อโรคยังไม่ถูกระบุ แต่การตรวจพบการกลายพันธุ์ของยีน TREX1 2) เป็นเบาะแสใหม่ที่บ่งชี้ถึงความเป็นไปได้ของการกลายพันธุ์ที่ไม่เกี่ยวข้องกับโครโมโซม X ด้วยการแพร่หลายของการวิเคราะห์จีโนมแบบครอบคลุม (WES และ WGS) คาดว่าการระบุยีนก่อโรคจะเร่งตัวขึ้น การตรวจสอบสมมติฐานที่เกี่ยวข้องกับ X และไม่เกี่ยวข้องกับ X เป็นภารกิจหลักในอนาคต
รายงานเกี่ยวกับชนิดแปรผันที่ไม่มีภาวะไม่มีคอร์ปัส คาโลซัมโดยสมบูรณ์ แต่มีเพียงการบางลง (dysgenesis) กำลังสะสมมากขึ้น 2) แม้ในกรณีที่ไม่เข้าเกณฑ์ทั้งสามข้อ การตรวจอวัยวะตาอย่างละเอียดและการใช้เกณฑ์การวินิจฉัยที่ขยายออกไปอาจช่วยเพิ่มความแม่นยำในการวินิจฉัย
Kang และคณะ (2022) รายงานการทำเลเซอร์โฟโตโคแอกกูเลชันและการตัดวุ้นตา 23G ในกรณีจอตาและวุ้นตา อักเสบสองข้างในกลุ่มอาการไอคาร์ดิ ซึ่งช่วยรักษาการเจริญเติบโตของลูกตาและปกป้องการทำงานของการมองเห็น 2) นอกจากนี้ยังมีการบันทึกว่ามีลาคูนาคอรอยด์ และจอตาใหม่ปรากฏให้เห็นหลังการผ่าตัด ซึ่งบ่งชี้ว่าการผ่าตัดตาสามารถช่วยยืนยันการวินิจฉัยได้เช่นกัน
มีการยอมรับเพิ่มขึ้นว่าการตรวจคัดกรองอวัยวะตาตั้งแต่เนิ่นๆ และการแทรกแซงอย่างรวดเร็วมีความสำคัญต่อการปกป้องการทำงานของจอตา 2) เลเซอร์ในบริเวณที่ไม่มีหลอดเลือดส่วนปลายมีศักยภาพในการป้องกันการลุกลามของจอตาพร่องออกซิเจน และหวังว่าจะมีการสะสมกรณีศึกษาเพิ่มเติมในอนาคต
การวิจัยเกี่ยวกับการใช้แคนนาบิไดออล (CBD) และการบำบัดด้วยอาหารคีโตเจนิกเพื่อควบคุมโรคลมชักกำลังดำเนินอยู่ 1) คาดว่าจะมีการสร้างทางเลือกการรักษาใหม่สำหรับโรคลมชักที่ดื้อต่อการรักษา
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCI D:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCI D:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.