Синдром Айкарди — редкое врожденное заболевание, впервые описанное в 1967 году французским неврологом Жаном Айкарди. Предполагается X-сцепленное доминантное наследование, поражающее почти исключительно девочек. У мальчиков он летален в гемизиготном состоянии, лишь несколько случаев описано у мальчиков с кариотипом XXY (синдром Клайнфельтера).
Частота оценивается примерно в 1:110 000 рождений, во всем мире насчитывается около 4 000 больных 1). Все случаи являются мутациями de novo, передачи от родителей к детям не наблюдается, риск рецидива у сибсов составляет менее 1% 1).
Классическая триада включает следующие три компонента 1):
Инфантильные спазмы: начало в возрасте около 3–4 месяцев.
Хориоретинальные лакуны (chorioretinal lacunae) : двусторонние круглые поражения глазного дна. Специфический признак данного заболевания.
Агенезия мозолистого тела (agenesis of the corpus callosum) : частичное или полное отсутствие.
Прогноз неблагоприятный. Средняя продолжительность жизни составляет 18 лет, а вероятность дожития до 27 лет оценивается в 0,62% 1).
QВозникает ли синдром Айкарди у мальчиков?
A
Это заболевание возникает почти исключительно у девочек. Предполагается X-сцепленное доминантное наследование, так как у мальчиков-гемизигот оно летально. Однако в мире зарегистрировано несколько случаев у мальчиков с кариотипом XXY (синдром Клайнфельтера).
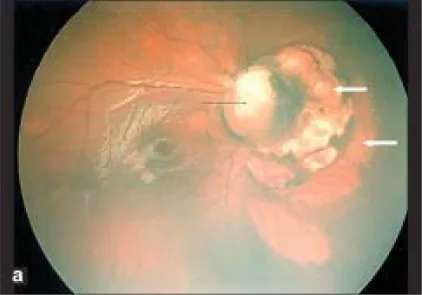
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Реткам-фото правого глаза показывает колобому диска зрительного нерва (черная стрелка) и куполообразные участки бледного цвета с четкими границами к носу от диска зрительного нерва, указывающие на хориоретинальные лакуны (белые стрелки).
Первым симптомом этого заболевания обычно являются инфантильные спазмы, появляющиеся в возрасте около 3-4 месяцев. Эпилепсия часто переходит в фармакорезистентную форму и сопровождается различными типами приступов.
Эпилептические приступы : начинаются с инфантильных спазмов и прогрессируют до фармакорезистентности. В одном случае в возрасте 4 месяцев появились генерализованные судороги 3-4 раза в день (продолжительностью 20-25 минут каждый) 1). В другом случае с 1 месяца наблюдались частые приступы мигания 2).
Психомоторная задержка развития : сопровождается тяжелой умственной отсталостью, часто затруднены самостоятельное передвижение и овладение речью.
Дисфункция желудочно-кишечного тракта : такие симптомы, как запор, присутствуют более чем в 90% случаев 1).
Нарушение зрения : обусловлено поражениями глазного дна, агенезией мозолистого тела и кортикальными мальформациями.
Среди офтальмологических находок данного заболевания хориоретинальные лакуны считаются патогномоничными.
Хориоретинальные лакуны
Распределение : двустороннее. Сгущение вокруг диска зрительного нерва и заднего полюса, но также распространяется на периферию.
Внешний вид : округлые или овальные поражения желтовато-белого или розового цвета. Распространенность 70–90%1).
Гистология : дефект пигментного эпителия сетчатки (ПЭС), простирающийся от сосудистой оболочки до обнаженной склеры2).
Течение : размер и количество могут увеличиваться со временем после операции2).
Другие глазные находки
Колобома зрительного нерва : встречается примерно в 44% случаев.
Микрофтальм : наблюдается примерно в 20% случаев.
Периферическая бессосудистая сетчатка : может образовываться бессосудистая зона на 360 градусов2).
Тракционная отслойка сетчатки (ТОС) : может возникать в сочетании с ножковой тканью (stalk tissue)2).
В описаниях клинических случаев зафиксирован пример с преретинальным кровоизлиянием и бессосудистой зоной на 360 градусов в правом глазу, а также ножковой тканью (фиброваскулярной ножкой) и тракционной отслойкой сетчатки в левом глазу2). Это также может быть причиной корковой зрительной недостаточности (CVI)3).
Агенезия мозолистого тела : частичное или полное отсутствие присутствует во всех случаях1). Истончение (дисгенезия) мозолистого тела также описано как вариант2).
Порок развития коры головного мозга: полимикрогирия (тип 2 по классификации Барковича), перивентрикулярные узелки серого вещества и многокамерные кисты выявляются на МРТ1).
Данные ЭЭГ: характерный паттерн с высокоамплитудными полиморфными ритмами и мультифокальными спайк-волновыми разрядами1).
Скелетные аномалии: у 40–60% отмечается сращение грудных позвонков (T9–T10) или позвонки-бабочки (T8)1).
QИзменяются ли хориоретинальные лакуны со временем?
A
Они могут прогрессировать. Сообщалось, что после офтальмологических хирургических вмешательств появляются новые лакуны, или их размер и количество увеличиваются со временем2). Важно регулярное наблюдение с помощью офтальмоскопии.
Синдром Айкарди предположительно наследуется по X-сцепленному доминантному типу, но ответственный ген на данный момент не идентифицирован1). Все случаи являются мутациями de novo, семейные случаи обычно не наблюдаются. Риск повторения для сибсов составляет менее 1%, и рекомендуется планирование следующего ребенка после генетического консультирования1).
В недавнем отчете о случае у одного пациента была обнаружена мутация гена TREX1 (c.292_293insA, p.(Cys99Metfs))2). TREX1 расположен на хромосоме 3, что частично противоречит гипотезе X-сцепленного наследования, и локализация ответственного гена продолжает обсуждаться.
Тип наследования: X-сцепленный доминантный (предположительно). Гемизиготные мужчины летальны внутриутробно.
Характер мутаций: все de novo. Наследственной передачи (от родителей к детям) нет.
Диагноз синдрома Айкарди в основном основывается на клиническом диагнозе, так как ответственный ген не идентифицирован. Подтверждение следующей классической триады является основой диагностики1).
Инфантильные спазмы (infantile spasms)
Хориоретинальные лакуны (chorioretinal lacunae)
Агенезия мозолистого тела (agenesis of the corpus callosum)
Даже если присутствуют только два из трех признаков, диагноз может быть поставлен в соответствии с расширенными диагностическими критериями, разработанными в 1999 году.
Состав расширенных диагностических критериев приведен ниже.
Мультидисциплинарное сотрудничество: для диагностики необходимо сотрудничество неврологии, офтальмологии, ортопедии и генетики 1).
QМожет ли генетическое тестирование подтвердить диагноз?
A
В настоящее время причинный ген для окончательного диагноза не идентифицирован, поэтому только генетическое тестирование не может подтвердить диагноз 1). Клинический диагноз основывается на сочетании клинических симптомов, данных офтальмоскопии и визуализации. Прогресс в области полногеномного анализа позволяет надеяться на выявление причинного гена в будущем.
Радикального лечения не существует. Цели лечения основаны на трех столпах: контроль эпилепсии, лечение офтальмологических осложнений и поддержка развития с помощью реабилитации.
Ведение эпилепсии
Препараты первой линии: фенитоин, леветирацетам, клобазам и др. 1).
Рефрактерные случаи: каннабидиол (КБД), кетогенная диета, каллозотомия, стимуляция блуждающего нерва могут быть опробованы 1).
Офтальмологическое вмешательство
Лазерная коагуляция: проводится на периферической бессосудистой сетчатке для предотвращения прогрессирования пролиферативной ретинопатии 2).
Витрэктомия: 23G витрэктомия при тракционной отслойке сетчатки (ТОС) 2).
Реабилитация
Раннее начало: начать физиотерапию, трудотерапию, логопедию и зрительную терапию сразу после постановки диагноза 1).
Уход при слабовидении: предоставление вспомогательных приспособлений и адаптация окружающей среды при нарушениях зрения.
Представлены примеры сообщений о хирургических вмешательствах при офтальмологических осложнениях.
Глаз
Находки
Лечение
Правый глаз
Преретинальное кровоизлияние, бессосудистая зона 360°
Лазерная фотокоагуляция
Левый глаз
Стеблевая ткань, ТРД
23G витрэктомия
После витрэктомии было подтверждено восстановление роста аксиальной длины: аксиальная длина левого глаза, составлявшая 17,45 мм в возрасте 1 месяца, выросла до 24,41 мм к 26 месяцам 2). Кроме того, сообщалось о случаях, когда после операции стали визуализироваться новые хориоретинальные лакуны, что способствовало подтверждению диагноза 2).
QВозможно ли офтальмологическое хирургическое лечение?
A
Да, возможно. Лазерная коагуляция периферической бессосудистой сетчатки и 23G-витрэктомия при тракционной отслойке сетчатки могут быть эффективны в некоторых случаях 2). Имеются сообщения об ускорении роста глаза после операции. Однако количество случаев невелико, и требуется осторожное ведение в специализированном учреждении.
Предполагается, что причиной этого заболевания является мутация de novo на X-хромосоме. Считается, что характер инактивации X-хромосомы (лионизация) приводит к фенотипическому разнообразию при одной и той же мутации. У мужчин мутация в гемизиготном состоянии летальна в эмбриональном периоде, выжить могут только мужчины с кариотипом XXY.
В последнее время у одного пациента с этим диагнозом была обнаружена мутация гена TREX1 (c.292_293insA, p.(Cys99Metfs)) 2). Поскольку TREX1 расположен на хромосоме 3, это может противоречить X-сцепленной гипотезе и указывать на возможность существования не-X-сцепленных генов-причин.
Гистологически хориоретинальные лакуны характеризуются дефектом пигментного эпителия сетчатки (ПЭС), простирающимся от хориокапилляров до обнаженной склеры 2). В области дефекта отсутствуют ПЭС и хориокапилляры, и наблюдается дисплазия недифференцированной нейросенсорной сетчатки.
Считается, что остатки персистирующей фетальной сосудистой системы (persistent fetal vasculature) участвуют в формировании фиброваскулярной ножки (stalk tissue) и периферической бессосудистой сетчатки 2). Эти аномальные сосуды вызывают тракционную отслойку сетчатки.
Кортикальное нарушение зрения (CVI) возникает как приобретенное снижение зрительной функции вследствие структурных аномалий головного мозга, таких как полимикрогирия, гетеротопия серого вещества и агенезия мозолистого тела 3).
Пороки развития головного мозга (полимикрогирия, агенезия мозолистого тела) основаны на нарушении миграции нейронов в эмбриональном периоде. Молекулярный механизм этого нарушения миграции остается невыясненным, и будущие исследования ожидаются вместе с идентификацией гена-причины.
7. Новейшие исследования и перспективы на будущее (отчёты на стадии исследований)
Ген-возбудитель остаётся неидентифицированным, однако обнаружение мутаций гена TREX1 2) является новой зацепкой, указывающей на возможное участие несцепленных с X-хромосомой мутаций. Распространение всестороннего геномного анализа (WES/WGS) должно ускорить идентификацию гена-возбудителя. Проверка гипотез сцепления с X-хромосомой и отсутствия сцепления является главной задачей на будущее.
Распознавание вариантных (атипичных) форм синдрома Айкарди
Накопляются сообщения о вариантных формах, при которых наблюдается не полное отсутствие мозолистого тела, а лишь его истончение (дисгенезия) 2). Даже в случаях, не удовлетворяющих всем трём критериям, детальное исследование глазного дна и применение расширенных диагностических критериев могут повысить точность диагностики.
Важность раннего офтальмологического вмешательства
Kang и соавт. (2022) сообщили о проведении лазерной фотокоагуляции и 23G-витрэктомии в случае двусторонней витреоретинопатии при синдроме Айкарди, что способствовало поддержанию роста глаза и защите зрительной функции 2). Они также отметили, что после операции стали визуализироваться новые хориоретинальные лакуны, что показывает, что офтальмологическая хирургия может способствовать и подтверждению диагноза.
Всё больше признаётся важность раннего скрининга глазного дна и быстрого вмешательства для защиты функции сетчатки 2). Лазерное воздействие на периферические бессосудистые зоны может предотвратить прогрессирование пролиферативной ретинопатии, и в будущем желательно накопление случаев.
Продвигаются исследования применения каннабидиола (CBD) и кетогенной диетотерапии для контроля эпилепсии 1). Ожидается создание новых вариантов лечения рефрактерной эпилепсии.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.