متلازمة أيكاردي (Aicardi syndrome) هي مرض خلقي نادر وصفه لأول مرة طبيب الأعصاب الفرنسي جان أيكاردي في عام 1967. يُفترض أنها وراثة سائدة مرتبطة بـ X، وجميع المرضى تقريبًا من الإناث. عند الذكور، تكون قاتلة في حالة نصف الزيجوت، لذلك تم الإبلاغ عن حالات قليلة فقط لدى الذكور ذوي النمط النووي XXY (متلازمة كلاينفلتر).
يقدر معدل الحدوث بحوالي 1:110,000 ولادة، ويقدر عدد المصابين في جميع أنحاء العالم بحوالي 4,000 شخص 1). جميع الحالات ناتجة عن طفرات جديدة (de novo)، ولا توجد حالات انتقال من الوالدين إلى الطفل، وخطر التكرار بين الأشقاء أقل من 1% 1).
تُعرف الثلاثية الكلاسيكية التالية 1):
تشنجات الرضع (infantile spasms): تبدأ في حوالي عمر 3-4 أشهر.
الثغرات المشيمية الشبكية (chorioretinal lacunae): آفات قاعية دائرية ثنائية الجانب. علامة مميزة لهذا المرض.
عدم تخلق الجسم الثفني (agenesis of the corpus callosum): غياب جزئي أو كامل.
الإنذار سيئ. متوسط العمر المتوقع هو 18 عامًا، وقد تم الإبلاغ عن احتمال البقاء على قيد الحياة حتى 27 عامًا بنسبة 0.62% 1).
Qهل تحدث متلازمة أيكاردي عند الذكور أيضًا؟
A
يحدث هذا المرض بشكل حصري تقريبًا عند الإناث. يُفترض أنه وراثة سائدة مرتبطة بـ X، حيث يكون مميتًا عند الذكور متماثلي الزيجوت. ومع ذلك، هناك عدد قليل من الحالات المبلغ عنها في جميع أنحاء العالم عند الذكور الذين لديهم النمط النووي XXY (متلازمة كلاينفلتر).
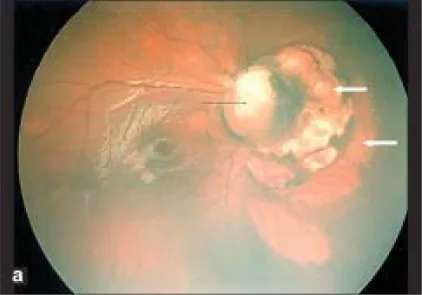
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
صورة Retcam للعين اليمنى تظهر ورمًا قولونيًا في القرص البصري (السهم الأسود) ومناطق مقببة شاحبة ذات حدود حادة أنفية للقرص البصري توحي بوجود ثغرات مشيمية شبكية (الأسهم البيضاء).
عادةً ما يكون العرض الأولي لهذا المرض هو الصرع الرمعي العضلي الطفلي (نوبات الرضاعة) الذي يظهر في عمر 3-4 أشهر. غالبًا ما يصبح الصرع مقاومًا للأدوية ويصاحبه أنواع نوبات متنوعة.
نوبات الصرع: تبدأ بنوبات الرضاعة وتتطور إلى مقاومة للأدوية. في إحدى الحالات، ظهرت تشنجات معممة 3-4 مرات يوميًا (تستمر 20-25 دقيقة لكل منها) في عمر 4 أشهر 1). في حالة أخرى، لوحظت نوبات رمش متكررة منذ عمر شهر واحد 2).
تأخر النمو النفسي الحركي: يصاحبه إعاقة ذهنية شديدة، وغالبًا ما يكون من الصعب تحقيق الحركة المستقلة أو اكتساب اللغة.
خلل وظيفي في الجهاز الهضمي: توجد أعراض هضمية مثل الإمساك في أكثر من 90% من الحالات 1).
ضعف البصر: يحدث ضعف البصر بسبب آفات قاع العين، وعدم تخلق الجسم الثفني، وتشوهات القشرة الدماغية.
شبكية لاوعائية محيطية: قد تتشكل منطقة لاوعائية تمتد 360 درجة2).
انفصال الشبكية الجرّي (TRD): قد يحدث مع نسيج الساق الوعائي (stalk tissue)2).
في تقارير الحالات، تم تسجيل حالة مع نزف تحت الشبكية في العين اليمنى ومنطقة لاوعائية محيطية 360 درجة، وفي العين اليسرى نسيج ساق وعائي (stalk tissue) وانفصال شبكية جرّي2). كما يمكن أن يسبب ضعفًا بصريًا قشريًا (cortical visual impairment; CVI)3).
غياب الجسم الثفني: غياب جزئي أو كامل موجود في جميع الحالات1). كما تم الإبلاغ عن ترقق الجسم الثفني (dysgenesis) كمتغير2).
تشوه القشرة المخية: يتم تأكيد وجود صغر التلفيف (polymicrogyria، النوع 2 حسب تصنيف Barkovich) والعقيدات الرمادية حول البطين والكيسات متعددة الحجرات بواسطة التصوير بالرنين المغناطيسي 1).
نتائج تخطيط كهربية الدماغ: يُظهر نمطًا مميزًا من الإيقاع متعدد الأشكال عالي الجهد مع موجات شوكية متعددة البؤر وتفريغات موجية 1).
تشوهات هيكلية: يُلاحظ في 40-60% من الحالات اندماج الفقرات الصدرية (T9-T10) والفقرات الفراشية (T8) وغيرها 1).
Qهل تتغير الثغرات المشيمية الشبكية بمرور الوقت؟
A
قد تتطور. تم الإبلاغ عن ظهور ثغرات جديدة بعد التدخل الجراحي للعين أو زيادة حجمها وعددها بمرور الوقت 2). من المهم إجراء متابعة منتظمة لفحص قاع العين.
يُفترض أن متلازمة إيكاردي هي وراثة سائدة مرتبطة بالكروموسوم X، ولكن لم يتم تحديد الجين المسبب حتى الآن 1). جميع الحالات هي طفرات جديدة (de novo)، ولا يُلاحظ حدوث حالات عائلية بشكل أساسي. خطر التكرار للأشقاء أقل من 1%، ويوصى بمناقشة التخطيط للطفل التالي بعد الاستشارة الوراثية1).
في تقارير الحالات الحديثة، تم اكتشاف طفرة في جين TREX1 (c.292_293insA، p.(Cys99Metfs)) في حالة واحدة 2). يقع TREX1 على الكروموسوم 3، مما يتعارض جزئيًا مع فرضية الارتباط بالكروموسوم X، ولا يزال الجدل قائمًا حول موقع الجين المسبب.
نمط الوراثة: سائد مرتبط بالكروموسوم X (مفترض). الذكور متماثلي الزيجوت يموتون في المرحلة الجنينية.
طبيعة الطفرة: جميعها طفرات جديدة (de novo). لا يوجد وراثة (انتقال من الوالدين إلى الطفل).
التعاون متعدد التخصصات: يجب أن يتعاون أطباء الأعصاب والعيون وجراحة العظام والوراثة في التشخيص 1).
Qهل يمكن تأكيد التشخيص بالاختبار الجيني؟
A
حاليًا، لم يتم تحديد جين مسبب يمكنه تأكيد التشخيص، لذلك لا يمكن الاعتماد على الاختبار الجيني وحده 1). يعتمد التشخيص بشكل أساسي على التقييم السريري الشامل للأعراض ونتائج فحص قاع العين والتصوير. مع تقدم التحليل الجيني الشامل، من المتوقع تحديد الجين المسبب في المستقبل.
بعد استئصال الزجاجية، لوحظ تعافي نمو طول المحور العيني؛ ففي العين اليسرى، نما طول المحور من 17.45 ملم عند شهر واحد بعد الولادة إلى 24.41 ملم عند 26 شهرًا2). كما تم الإبلاغ عن حالات أصبحت فيها الثغرات المشيمية الشبكية مرئية بعد الجراحة، مما ساعد في تأكيد التشخيص2).
Qهل العلاج الجراحي للعين ممكن؟
A
نعم، ممكن. قد يكون التخثير الضوئي بالليزر للشبكية اللاوعائية المحيطية واستئصال الزجاجية 23G لانفصال الشبكية الجري فعالين في بعض الحالات2). هناك تقارير عن تسارع نمو العين بعد الجراحة. ومع ذلك، فإن عدد الحالات محدود، ويتطلب الأمر تعاملًا حذرًا في المراكز المتخصصة.
يُعتقد أن الطفرات الجديدة على الكروموسوم X هي سبب هذا المرض. يُعتقد أن نمط تعطيل الكروموسوم X (تأين) يؤدي إلى تنوع في النمط الظاهري حتى مع نفس الطفرة. في الذكور، تكون الطفرة متماثلة الزيجوت، مما يؤدي إلى الوفاة في المرحلة الجنينية، ولا يمكن البقاء على قيد الحياة إلا للذكور الذين لديهم النمط النووي XXY.
في السنوات الأخيرة، تم اكتشاف طفرة في جين TREX1 (c.292_293insA, p.(Cys99Metfs)) في حالة واحدة تم تشخيصها بهذا المرض2). نظرًا لأن TREX1 يقع على الكروموسوم 3، فقد يتعارض ذلك مع فرضية الارتباط بالكروموسوم X، مما يشير إلى احتمال وجود جين مسبب غير مرتبط بـ X.
كسمة نسيجية للثغرات المشيمية الشبكية، تم التأكد من أن فقدان الظهارة الصبغية للشبكية (RPE) يمتد من الصفيحة الشعرية المشيمية إلى طبقة الصلبة العارية2). لا توجد الظهارة الصبغية للشبكية والصفيحة الشعرية المشيمية في موقع الخلل، ويلاحظ خلل تنسج في الشبكية العصبية غير المتمايزة.
يُعتقد أن بقايا الجهاز الوعائي الجنيني المستمر (persistent fetal vasculature) تشارك في تكوين الساق الليفي الوعائي (stalk tissue) والشبكية اللاوعائية المحيطية2). تسبب هذه الأوعية الدموية غير الطبيعية انفصال الشبكية الجري.
الاضطراب البصري القشري (CVI) هو انخفاض مكتسب في الوظيفة البصرية ناتج عن تشوهات هيكلية في الدماغ مثل صغر التلافيف (polymicrogyria) والمادة الرمادية المنتبذة (heterotopic gray matter) وغياب الجسم الثفني3).
تشوهات الدماغ (صغر التلافيف، غياب الجسم الثفني) تستند إلى اضطراب هجرة الخلايا العصبية في المرحلة الجنينية. لا تزال الآلية الجزيئية المسببة لاضطراب الهجرة هذا غير معروفة، ويُنتظر البحث المستقبلي مع تحديد الجين المسبب.
7. أحدث الأبحاث والتوجهات المستقبلية (تقارير المرحلة البحثية)
لا يزال الجين المسبب غير محدد، لكن اكتشاف طفرة جين TREX1 2) هو دليل جديد يشير إلى احتمال تورط طفرات غير مرتبطة بالكروموسوم X. من المتوقع أن يؤدي انتشار التحليل الجيني الشامل (WES وWGS) إلى تسريع تحديد الجين المسبب. التحقق من فرضية الارتباط بالكروموسوم X والفرضية غير المرتبطة به هو مهمة رئيسية في المستقبل.
تتراكم التقارير عن الحالات المتحولة التي لا تظهر غيابًا كاملاً للجسم الثفني بل ترققًا فقط (dysgenesis) 2). حتى في الحالات التي لا تستوفي جميع المعايير الثلاثة، يمكن أن يؤدي تطبيق فحص قاع العين التفصيلي ومعايير التشخيص الموسعة إلى تحسين دقة التشخيص.
أبلغ كانغ وآخرون (2022) عن إجراء تخثير ضوئي بالليزر واستئصال الزجاجية 23G لحالة اعتلال الشبكية الزجاجي الثنائي في متلازمة أكاردي، مما ساهم في الحفاظ على نمو العين وحماية الوظيفة البصرية 2). كما تم تسجيل ظهور ثغرات مشيمية شبكية جديدة بعد الجراحة، مما يشير إلى أن الجراحة العينية يمكن أن تساهم أيضًا في تأكيد التشخيص.
هناك اعتراف متزايد بأهمية فحص قاع العين المبكر والتدخل السريع لحماية وظيفة الشبكية2). قد يمنع الليزر على المناطق اللاوعائية المحيطية تطور اعتلال الشبكية التكاثري، ويأمل في تجميع المزيد من الحالات في المستقبل.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
انسخ نص المقال والصقه في مساعد الذكاء الاصطناعي الذي تفضله.
تم نسخ المقال إلى الحافظة
افتح أحد مساعدي الذكاء الاصطناعي أدناه والصق النص المنسوخ في مربع المحادثة.