Iride
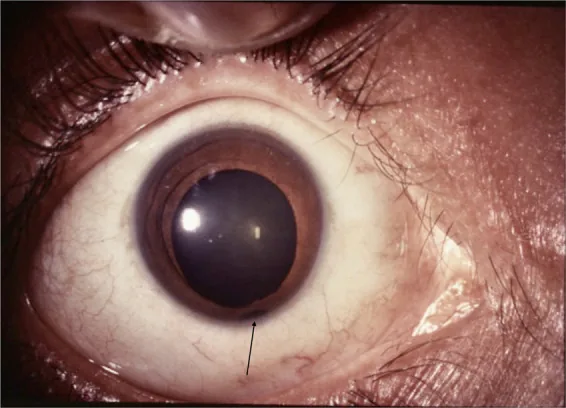
Pupilla a buco di serratura : tipicamente il difetto è situato in basso e verso il naso, deformando la pupilla a forma di buco di serratura.
Localizzazione infero-temporale : può verificarsi anche in sedi atipiche.

Il coloboma (coloboma) deriva dalla parola greca che significa «difetto» ed è una malattia congenita caratterizzata da difetti tissutali in varie parti dell’occhio a causa di una chiusura incompleta della fessura embrionale. Può verificarsi a livello di palpebre, iride, cristallino, corpo ciliare, coroide, retina e nervo ottico. Il difetto è tipicamente localizzato in sede infero-nasale ed è spesso associato a microftalmia.
La prevalenza è stimata tra 0,5 e 2,2 casi ogni 10.000 nati. Negli Stati Uniti è di circa 2,6 casi ogni 10.000 nati 4), mentre in Europa è riportata tra 4 e 19 casi ogni 100.000 nati 6). Rappresenta circa l’11% della cecità infantile e il tasso di diagnosi genetica è inferiore al 30% 6). La prevalenza del coloboma palpebrale è di 0,2-0,8 casi ogni 10.000 nati. Costituisce lo 0,07% delle malformazioni oculari congenite e il 3,2-11,2% nei bambini ipovedenti.
Esistono colobomi tipici e atipici. I colobomi tipici derivano da una chiusura incompleta della fessura embrionale e si trovano nel quadrante infero-nasale, mentre i colobomi atipici si verificano in altre sedi e implicano diversi meccanismi di sviluppo.
I codici ICD-10 sono Q10.3 (palpebra), Q13.0 (iride), Q12.2 (cristallino), H47.319 (nervo ottico) e Q14.8 (coroide/retina).
Esistono sia forme sporadiche che ereditarie. Sono state riportate diverse modalità di trasmissione, tra cui autosomica dominante, autosomica recessiva e legata all’X. Sono stati identificati diversi geni causali, come PAX2, CHD7 e FZD5, ma il tasso di diagnosi genetica è inferiore al 30% 6). In caso di storia familiare, si raccomanda la consulenza genetica.
L’acuità visiva varia ampiamente, dall’assenza di percezione luminosa alla normalità, a seconda della sede e dell’estensione del difetto.
Il coloboma presenta reperti caratteristici in ogni parte dell’occhio.
Iride
Pupilla a buco di serratura : tipicamente il difetto è situato in basso e verso il naso, deformando la pupilla a forma di buco di serratura.
Localizzazione infero-temporale : può verificarsi anche in sedi atipiche.
Corioide e retina
Lesioni giallo-biancastre : difetto circolare o a ventaglio, dai bordi netti, attraverso cui traspare la sclera.
Rischio di distacco di retina : incidenza del 23-40%7). Necessario follow-up regolare.
Nervo ottico e cristallino
Aumento dell’escavazione del nervo ottico : da unilaterale a bilaterale, di grado variabile.
Appiattimento dell’equatore del cristallino : dovuto a un difetto della zonula di Zinn. Osservabile sotto midriasi.
Palpebra
Difetto interno della palpebra superiore : difetto tissutale a tutto spessore.
Associazione con malformazioni sistemiche : talvolta isolato, ma può essere associato ad anomalie sistemiche.
Il coloboma ciliare isolato è raro; spesso si riscontra in continuità con un grande coloboma coroideale.
L’acuità visiva varia dall’assenza di percezione luminosa alla normalità. Se il coloboma è limitato all’iride, la vista è spesso preservata. Se coinvolge la macula o il nervo ottico, la vista tende a essere scarsa.
La causa principale del coloboma è la chiusura incompleta della fessura embrionale.
La fessura embrionale (fessura ottica) si forma alla 4a settimana di gestazione e si completa alla 5a settimana. La chiusura inizia alla 6a settimana e termina alla 7a settimana. Se questo processo di chiusura viene interrotto per qualsiasi motivo, si sviluppa un coloboma. È stato anche suggerito il coinvolgimento della vitamina A.
Sono stati identificati diversi geni coinvolti nello sviluppo del coloboma.
| Gene | Malattia/fenotipo associato |
|---|---|
| PAX2 | Sindrome renale-coloboma5) |
| CHD7 | Sindrome CHARGE |
| FZD5 | Coloboma oculare sintomatico + microcornea6) |
| TENM3 | MCOPS15 (microcornea + ritardo dello sviluppo) 8) |
| FAT1 | Coloboma + nefropatia 9) |
| YAP1 | Associato a coloboma |
| ABCB6 | Associato a coloboma |
| SALL2 | Associato a coloboma |
Il coloboma può associarsi alle seguenti sindromi sistemiche.
È una sindrome da malformazioni multiple causata da una mutazione del gene CHD7. Il nome deriva dalle iniziali di coloboma (C), cardiopatia (H), atresia delle coane (A), ritardo di crescita e sviluppo (R), ipoplasia genitale (G) e anomalie dell’orecchio (E). La diagnosi si basa sulla combinazione di questi segni.
Vengono eseguiti test genetici completi come il sequenziamento dell’esoma (WES), ma il tasso diagnostico rimane inferiore al 30%6).
Il coloboma richiede una diagnosi differenziale con le seguenti patologie in base alla sede.
| Sede | Principali diagnosi differenziali |
|---|---|
| Palpebra | Sindrome da bande amniotiche, trauma |
| Iride | Aniridia, dialisi traumatica dell’iride |
| Nervo ottico | Sindrome del morning glory, ipoplasia del nervo ottico |
Non esiste un trattamento curativo per il coloboma; la gestione si basa su terapia sintomatica e controllo delle complicanze in base alla sede.
Castilla-Martinez et al. (2024) hanno eseguito un intervento di cataratta con laser a femtosecondi (FLACS) combinato a pupilloplastica e posizionamento di CTR in un caso di coloboma di iride, cristallino e zonula associato a cataratta. L’acuità visiva postoperatoria è migliorata a logMAR 0,24).
Nel coloboma del nervo ottico, a causa dell’ipoplasia della lamina cribrosa, l’arteria e la vena centrali della retina si dividono già posteriormente alla papilla, e i vasi retinici originano da più siti al margine della papilla. Al di sotto della papilla si osserva spesso atrofia corioretinica dovuta a chiusura incompleta della fessura embrionale.
Per il distacco regmatogeno della retina si esegue una vitrectomia. Sono state riportate tecniche chirurgiche come il riattacco retinico con colla di fibrina 7) e la fotocoagulazione endoculare con tamponamento a gas 3). Nel distacco sieroso può verificarsi una regressione spontanea, e la strategia terapeutica viene decisa individualmente.
La coppa ottica si forma dal neuroectoderma alla 4a settimana di gestazione. Sulla faccia ventrale della coppa ottica si forma una fessura embrionale (fessura ottica), attraverso cui passa l’arteria ialoidea. Questa fessura si completa alla 5a settimana e inizia a chiudersi dalla 6a settimana. La chiusura inizia vicino all’equatore e procede anteriormente (verso l’iride) e posteriormente (verso il nervo ottico), completandosi alla 7a settimana.
Il processo di chiusura coinvolge la transizione epitelio-mesenchimale (EMT). Le cellule epiteliali della retina neurale al bordo della fessura embrionale degradano la membrana basale, acquisiscono un fenotipo mesenchimale e si fondono. Un’alterazione di questo processo porta alla formazione di un coloboma.
Il gene FZD5 codifica per un recettore della via di segnalazione Wnt. Le mutazioni ipofunzionali di FZD5 causano un’anomalia della segnalazione Wnt, portando a un difetto di chiusura della fessura embrionale e a microcornea 6).
Anche le cellule della cresta neurale (NCC) sono coinvolte nella genesi del coloboma. Le NCC si differenziano in tessuto mesenchimale intorno alla coppa ottica e svolgono un ruolo importante nel processo di chiusura della fessura embrionale 2). Un’alterazione della migrazione delle NCC porta ad anomalie dello sviluppo dell’iride e della coroide.
Cortes-Gonzalez et al. (2024) hanno riportato che una mutazione missenso omozigote di FZD5 (p.M160V) causa coloboma oculare sintomatico e microcornea 6). Mostra un modello di ereditarietà recessiva e l’analisi funzionale ha confermato un’alterazione dell’attivazione ligando-dipendente della via di segnalazione Wnt. Il tasso di diagnosi genetica del coloboma è inferiore al 30% e si prevede che l’identificazione di nuovi geni causali migliori la diagnosi.
Zhou et al. (2022) hanno riportato che mutazioni eterozigoti composte del gene TENM3 causano MCOPS15 (microcornea, coloboma irido-coroideo, ritardo globale dello sviluppo) 8). TENM3 codifica una proteina transmembrana coinvolta nell’adesione cellulare e nella neurogenesi.
Esmaeilzadeh et al. (2022) hanno riportato l’identificazione di mutazioni del gene FAT1 in una famiglia iraniana con coloboma dell’iride e nefropatia 9). FAT1 è un membro della superfamiglia delle caderine coinvolto nella polarità cellulare e nella morfogenesi tissutale.
Hu et al. (2024) hanno riportato l’identificazione di una mutazione frameshift c.76delG di PAX2 in una famiglia con glomerulosclerosi segmentaria e focale (FSGS) 5). Questo risultato suggerisce che lo spettro fenotipico della sindrome del coloboma renale sia più ampio di quanto si pensasse in precedenza.
Jain et al. (2024) hanno riportato un caso di distacco di retina associato a coloboma trattato con retinopessia con colla di fibrina 7). La tecnica prevede l’applicazione di colla di fibrina attorno alla lacerazione retinica al margine del coloboma per rafforzare l’adesione; l’acuità visiva finale è migliorata a 20/50.
Ratra et al. (2023) hanno riportato un caso di coloboma coroideo atipico complicato da fistola sclerale post-traumatica, trattato con successo con vitrectomia, fotocoagulazione endoculare e tamponamento gassoso 3).
Scemla et al. (2021) hanno riportato il caso di un uomo di 19 anni con filtrazione transclerale in sede di coloboma coroideo, che ha causato ipotonia (4 mmHg) 1). La microscopia ultrasonica ha confermato un difetto sclerale. Il recupero spontaneo è avvenuto in 6 settimane, con pressione intraoculare di 11 mmHg e acuità visiva di 1,0.