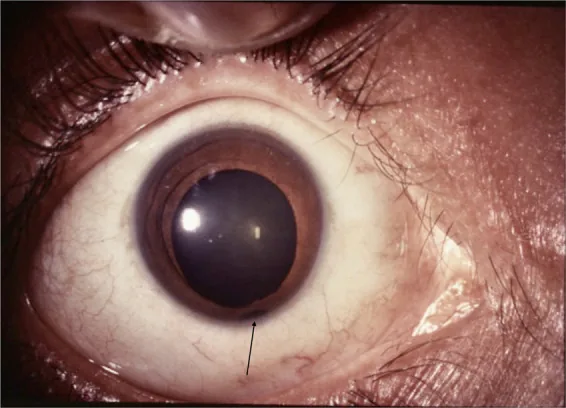
İris
Anahtar deliği şeklinde pupil: Tipik olarak nazal alt kadranda yer alan defekt, pupili anahtar deliği şeklinde deforme eder.
Temporal alt kadrana doğru: Atipik bir konumda da ortaya çıkabilir.

Kolobom, Yunanca “eksiklik” anlamına gelen bir kelimeden türemiştir ve fetal yarığın kapanmaması sonucu gözün çeşitli bölgelerinde doku eksikliği ile karakterize konjenital bir hastalıktır. Göz kapağı, iris, lens, siliyer cisim, koroid, retina ve optik sinirde görülebilir. Tipik olarak nazal-alt kadranda yer alır ve sıklıkla mikrof talmi eşlik eder.
Prevalansı 10.000 doğumda 0,5-2,2 vaka olarak bildirilmiştir. ABD’de 10.000 doğumda yaklaşık 2,6 vaka 4), Avrupa’da ise 100.000 doğumda 4-19 vaka rapor edilmiştir 6). Çocukluk çağı körlüğünün yaklaşık %11’ini oluşturur ve genetik tanı oranı %30’un altındadır 6). Göz kapağı kolobomunun prevalansı 10.000 doğumda 0,2-0,8 vakadır. Konjenital göz anomalileri içindeki oranı %0,07, görme engelli çocuklarda ise %3,2-11,2 olarak bildirilmiştir.
Kolobomlar tipik ve atipik olarak ikiye ayrılır. Tipik kolobomlar fetal yarığın kapanmamasına bağlı olup nazal-alt kadranda yer alırken, atipik kolobomlar diğer bölgelerde görülür ve farklı bir gelişim mekanizması olduğu düşünülür.
ICD-10 kodları: Q10.3 (göz kapağı), Q13.0 (iris), Q12.2 (lens), H47.319 (optik sinir), Q14.8 (koroid ve retina).
Hem sporadik hem de kalıtsal formları vardır. Otozomal dominant, otozomal resesif ve X’e bağlı gibi çeşitli kalıtım şekilleri bildirilmiştir. PAX2, CHD7, FZD5 gibi birden fazla nedensel gen tanımlanmış olmasına rağmen genetik tanı oranı %30’un altındadır6). Aile öyküsü varsa genetik danışmanlık önerilir.
Görme keskinliği, defektin yeri ve boyutuna bağlı olarak ışık hissi yokluğundan normale kadar geniş ölçüde değişir.
Kolobom, gözün her bir bölgesinde karakteristik bulgular gösterir.
İris
Anahtar deliği şeklinde pupil: Tipik olarak nazal alt kadranda yer alan defekt, pupili anahtar deliği şeklinde deforme eder.
Temporal alt kadrana doğru: Atipik bir konumda da ortaya çıkabilir.
Koroid ve Retina
Sarı-beyaz lezyonlar: Skleranın görülebildiği, sınırları belirgin yuvarlak veya yelpaze şeklinde defektler.
Retina dekolmanı riski: Görülme sıklığı %23-407). Düzenli takip gereklidir.
Optik Sinir ve Lens
Optik disk çukurlaşmasında genişleme: Tek taraflıdan çift taraflıya kadar değişen derecelerde.
Lens ekvatorunda düzleşme: Zinn zonüllerindeki defekte bağlı oluşur. Pupil genişlemesi altında gözlenir.
Göz Kapağı
Üst göz kapağında iç tarafta defekt: Tam kat doku kaybı.
Sistemik anomalilerle birliktelik: İzole olgular olmakla birlikte sıklıkla sistemik anomaliler eşlik eder.
Siliyer cisim kolobomu nadiren izole olarak görülür ve genellikle büyük bir koroid kolobomu ile bitişik olarak bulunur.
Görme keskinliği ışık hissi yokluğundan normale kadar geniş bir aralıkta değişir. Yalnızca irisi etkileyen sınırlı bir kolobomda görme genellikle korunur. Makula veya optik sinir tutulumunda görme kaybı daha sıktır.
Kolobomun ana nedeni fetal fissürün kapanma kusurudur.
Fetal fissür (optik fissür) gebeliğin 4. haftasında oluşur ve 5. haftada tamamlanır. Kapanma 6. haftada başlar ve 7. haftada sonlanır. Bu kapanma sürecinin herhangi bir nedenle bozulması koloboma yol açar. A vitamini eksikliğinin de rol oynadığı belirtilmektedir.
Kolobom oluşumunda rol oynayan birden fazla gen tanımlanmıştır.
| Gen | İlgili Hastalık/Fenotip |
|---|---|
| PAX2 | Renal kolobom sendromu5) |
| CHD7 | CHARGE sendromu |
| FZD5 | Semptomatik OK + mikrokornea 6) |
| TENM3 | MCOPS15 (mikrokornea + gelişim geriliği) 8) |
| FAT1 | Kolobom + nefropati 9) |
| YAP1 | Kolobom ile ilişkili |
| ABCB6 | Kolobom ile ilişkili |
| SALL2 | Kolobom ile ilişkili |
Kolobom aşağıdaki sistemik sendromlarla birlikte görülebilir.
CHD7 gen mutasyonu sonucu oluşan çoklu malformasyon sendromudur. Kolobom (C), kalp hastalığı (H), koanal atrezi (A), büyüme ve gelişme geriliği (R), genital hipoplazi (G), kulak anomalisi (E) baş harflerinden oluşan isim, bulguların kombinasyonu ile tanı konur.
Tüm ekzom dizileme (WES) gibi kapsamlı genetik testler yapılır, ancak tanı oranı %30’un altında kalır6).
Kolobom, yerleşim yerine göre aşağıdaki hastalıklarla ayırıcı tanı gerektirir.
| Yerleşim | Başlıca ayırıcı tanılar |
|---|---|
| Göz kapağı | Amniyotik bant sendromu, travma |
| İris | Aniridi, travmatik iris diyalizi |
| Optik sinir | Sabah zaferi sendromu, optik sinir hipoplazisi |
Koloboma için kesin bir tedavi yoktur; tedavi, etkilenen bölgeye göre semptomatik ve komplikasyon yönetimine odaklanır.
Castilla-Martinez ve ark. (2024), iris, lens ve zonüler kolobom ile birlikte kataraktı olan bir olguda femtosaniye lazer katarakt cerrahisi (FLACS) ve pupilloplasti uygulayarak CTR yerleştirdi. Ameliyat sonrası görme keskinliği logMAR 0.2’ye iyileşti4).
Optik sinir kolobomunda, lamina kribrozada hipoplazi olduğu için retina santral arter ve veni papilla arkasında dallanır ve retina damarları papilla kenarında birden fazla noktadan başlar. Papilla altında, fetal fissür kapanma defektine bağlı koroidoretinal atrofi sıklıkla görülür.
Regmatojen retina dekolmanında vitrektomi yapılır. Fibrin yapıştırıcı ile kombine retina restorasyonu7) ve endolaser fotokoagülasyon + gaz tamponadı3) gibi cerrahi teknikler bildirilmiştir. Seröz retina dekolmanında kendiliğinden gerileme görülebileceğinden tedavi yaklaşımı bireysel olarak belirlenir.
Optik kadeh, embriyonik 4. haftada nöroektodermden oluşur. Optik kadehin ventral tarafında, vitreus arterinin geçtiği embriyonik yarık (optik kadeh yarığı) oluşur. Bu yarık 5. haftada tamamlanır ve 6. haftadan itibaren kapanmaya başlar. Kapanma ekvator bölgesinden başlayarak öne (iris tarafı) ve arkaya (optik sinir tarafı) doğru ilerler ve 7. haftada tamamlanır.
Kapanma sürecinde epitelyal-mezenkimal geçiş (EMT) rol oynar. Embriyonik yarık kenarındaki nöral retina epitel hücreleri bazal membranı parçalar, mezenkimal fenotip kazanır ve kaynaşır. Bu sürecin bozulması koloboma yol açar.
FZD5 geni, Wnt sinyal yolunun reseptörünü kodlar. FZD5’in işlev kaybı mutasyonları, Wnt sinyalinde anormalliğe yol açarak embriyonik yarık kapanma kusuruna ve mikrokorneaya neden olur6).
Nöral krest hücreleri (NCC) de kolobom oluşumunda rol oynar. NCC, optik kadeh çevresindeki mezenkimal dokuya farklılaşır ve embriyonik yarığın kapanma sürecinde önemli bir rol oynar2). NCC göçünün bozulması, iris ve koroid gelişim anormalliklerine yol açar.
Cortes-Gonzalez ve ark. (2024), FZD5’teki homozigot yanlış anlamlı mutasyonun (p.M160V) semptomatik oküler kolobom ve mikrokorreaya neden olduğunu bildirmiştir6). Resesif kalıtım paterni gösterir ve fonksiyonel analizler, Wnt sinyal yolağının ligand bağımlı aktivasyonunun bozulduğunu doğrulamıştır. Kolobomda genetik tanı oranı %30’un altındadır ve yeni nedensel genlerin tanımlanmasının tanıyı iyileştirmeye katkıda bulunması beklenmektedir.
Zhou ve ark. (2022), TENM3 genindeki birleşik heterozigot mutasyonların MCOPS15’e (mikrokornea, iris-koroid kolobomu, yaygın gelişimsel gecikme) neden olduğunu bildirmiştir8). TENM3, hücre adezyonu ve nörogelişimde rol oynayan bir transmembran proteini kodlar.
Esmaeilzadeh ve ark. (2022), FAT1 gen mutasyonlarının iris kolobomu ve nefropati ile seyreden bir İran ailesinde tanımlandığını bildirmiştir9). FAT1, hücre polaritesi ve doku morfogenezinde rol oynayan kadherin süper ailesinin bir üyesidir.
Hu ve ark. (2024), PAX2 genindeki c.76delG çerçeve kayması mutasyonunun fokal segmental glomerüloskleroz (FSGS) olan bir ailede tanımlandığını bildirmiştir5). Bu bulgu, renal kolobom sendromunun fenotipik spektrumunun daha önce düşünülenden daha geniş olduğunu göstermektedir.
Jain ve ark. (2024), kolobom ilişkili retina dekolmanı için fibrin yapıştırıcı ile kombine retina restorasyonu uygulanan bir olgu bildirmiştir7). Kolobom kenarındaki retina yırtığı çevresine fibrin yapıştırıcı sürülerek yapışma güçlendirilmiş ve son görme keskinliği 20/50’ye yükselmiştir.
Ratra ve ark. (2023), atipik koroid kolobomu ile travma sonrası skleral fistülün eşlik ettiği bir olguda vitrektomi, endolaser fotokoagülasyon ve gaz tamponadı ile başarılı tedavi bildirmiştir3).
Scemla ve ark. (2021), 19 yaşında bir erkek hastanın koroidal koloboma bölgesinde transskleral filtrasyon gelişmesi sonucu hipotoni (4 mmHg) oluşan bir olgu bildirdi1). Ultrasonik biyomikroskopi ile skleral defekt doğrulandı. Altı haftada kendiliğinden iyileşti, göz içi basıncı 11 mmHg ve görme keskinliği 1.0 olarak korundu.