İzole Aniridi
Sıklık: Toplamın yaklaşık 2/3’ü.
Kalıtım şekli: Otozomal dominant (AD).
Özellikler: PAX6 gen mutasyonuna bağlıdır. Sistemik bulgu yoktur. Penetrans tamdır ancak ekspresyon değişkendir.

Aniridi, irisin değişen derecelerde az gelişmişliği veya yokluğu ile karakterize nadir bir konjenital hastalıktır. “Aniridi” terimi bir yanlış adlandırmadır; goniyoskopi veya ultrasonik biyomikroskopi (UBM) ile hemen her zaman iris dokusu parçaları tespit edilebilir.
Prevalansın yaklaşık 1/40.000 ila 1/100.000 olduğu ve belirgin bir ırk veya cinsiyet farkı bildirilmediği belirtilmektedir1). ICD-10’da Q13.1 olarak sınıflandırılır.
Aniridi, sadece irisi değil aynı zamanda kornea, lens, açı, fovea ve optik siniri de etkileyen panoküler bir hastalıktır1) ve görmeyi tehdit eden çeşitli oküler komplikasyonlara yol açar. Görme prognozu genellikle kötüdür ve çoğu durumda düzeltilmiş görme keskinliği 0,1 civarında kalır. Pupil refleksi kaybolmuştur ancak akomodasyon refleksi korunur; vakaların %60-90’ı bilateraldir.
Aşağıdaki üç fenotip tanınmaktadır.
İzole Aniridi
Sıklık: Toplamın yaklaşık 2/3’ü.
Kalıtım şekli: Otozomal dominant (AD).
Özellikler: PAX6 gen mutasyonuna bağlıdır. Sistemik bulgu yoktur. Penetrans tamdır ancak ekspresyon değişkendir.
WAGR Sendromu
Sıklık: Sporadik vakaların bir kısmı.
Kalıtım şekli: PAX6 ve WT1’in bitişik delesyonu.
Özellikler: Wilms tümörü, genitoüriner anomaliler ve zihinsel gerilik eşlik eder. Tümör riski en fazla %50’dir.
Gillespie Sendromu
Sıklık: Toplamın yaklaşık %2’si.
Kalıtım şekli: ITPR1 gen mutasyonu.
Özellikler: Serebellar ataksi ve zihinsel engellilik eşlik eder. Sabit midriyazis gösteren spesifik iris anomalisi karakteristiktir3).
Sporadik aniridi, toplamın yaklaşık 1/3’ünü oluşturur ve PAX6 dahil 11p13’ün de novo delesyonu sonucu oluşur. Bitişik WT1 genine kadar uzanan delesyon WAGR sendromuna neden olur1). Sporadik aniridili hastaların %25-30’unda Wilms tümörü gelişir ve rölatif risk 67 olarak bildirilmiştir.
PAX6, göz gelişiminin ana kontrol genidir ve göz, nöral tüp, olfaktör bulb, pankreas Langerhans adacıkları ve olfaktör epitelin gelişiminde rol oynar. Tek alel kaybı (haploinsuffiency) ile ortaya çıkar; her iki alel anormal olduğunda embriyonik ölümle sonuçlanır. 2017 yılında Nadir Hastalıklar Yasası kapsamında belirlenmiş nadir hastalık haline gelmiş olup, şiddet derecesi III ve üzeri (tanı ve test bölümüne bakınız) olan hastalar tıbbi masraf desteğinden yararlanabilir7).
Sporadik (yeni mutasyon) vakalar tüm vakaların yaklaşık 1/3’ünü oluşturur ve aile öyküsü olmadan da ortaya çıkabilir. Sporadik vakalarda WAGR sendromu olasılığı nedeniyle genetik test ve abdominal ultrason ile Wilms tümörü taraması önemlidir.
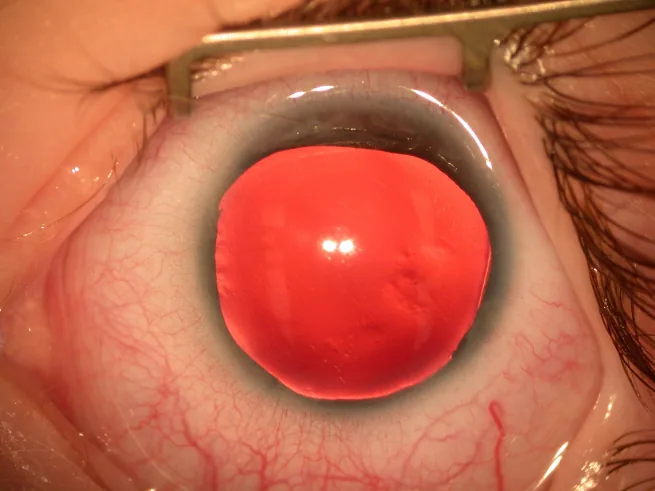
Aniridi vakalarının çoğu doğumda iris/pupil anormalliği veya bebeklik döneminde nistagmus ile tespit edilir.
Fenotip aileler arasında ve aile içinde farklılık gösterebilir, ancak sağ ve sol göz arasındaki fark genellikle küçüktür.
Başlıca neden foveal hipoplazi olup, düzeltilmiş görme keskinliği genellikle 0,1-0,2 civarındadır. Maküler hipoplazi eşlik ettiğinde görme prognozu özellikle kötüdür. Bebeklik ve çocukluk döneminden itibaren refraksiyon düzeltmesi ve az görme rehabilitasyonu görsel gelişim için önemlidir.
PAX6, göz dokusunun yanı sıra merkezi sinir sistemi, pankreas Langerhans adacıkları ve olfaktör epitelde de eksprese edildiğinden, aşağıdaki göz dışı komplikasyonlar görülebilir8).
Görme fonksiyonunu belirleyen önemli faktörler glokom, maküler hipoplazi, nistagmus, keratopati, katarakt ve iris displazisidir. Glokoma bağlı görme alanı ve görme keskinliği kaybı geri dönüşümsüz olduğundan, takipte göz içi basıncı yönetimi en önemli unsurdur 8).
Konjenital aniridi vakalarının çoğu, kromozom 11’in kısa kolunda (11p13) yer alan PAX6 genindeki heterozigot mutasyonlardan kaynaklanır. PAX6’da haployetmezlik ana patojenik mekanizmadır 1).
PAX6 geni, göz gelişiminin ana kontrol genidir ve göz, nöral tüp, koku soğanı ve pankreas gelişiminde kritik rol oynar. Normal göz gelişimi için iki kopya PAX6 gereklidir ve bir kopyanın işlev kaybı aniridiye yol açar 1).
Çinli hastalarla yapılan bir kohort çalışmasında, vakaların %96,9’unda PAX6 geninde nedensel mutasyonlar tespit edilmiştir 1). Tipik aniridide, vakaların %96’sında nonsense aracılı mRNA yıkımını (NMD) indükleyen mutasyonlar veya büyük delesyonlar saptanır 1).
Patolojik olarak, iris kökü korunurken düz kas eksikliği ve açı gelişim bozukluğu görülür. Korneal epitel kök hücre fonksiyon bozukluğu, epitel ve Bowman membranında anormalliklere ve damardan zengin pannus oluşumuna yol açar.
Aniridi fenotipine neden olan PAX6 mutasyonlarının dağılımı aşağıda gösterilmiştir.
| Mutasyon Tipi | Sıklık |
|---|---|
| Anlamsız mutasyon | Yaklaşık %39 |
| Çerçeve kayması mutasyonu | Yaklaşık %25 |
| Splice bölgesi mutasyonu | Yaklaşık %13 |
| Yanlış anlamlı mutasyon | Yaklaşık %12 |
Run-on mutasyonları (C-terminal uzama mutasyonları) yaklaşık %5’ini oluşturur ve durdurma kodonunun bir translasyon kodonuna dönüşmesiyle anormal şekilde uzamış PAX6 proteini üretilir6). C-terminal uzama mutasyonları sıklıkla şiddetli iris hipoplazisi ve ileri düzeyde görme bozukluğu ile birlikte görülür1)6).
Gen mutasyonları çoğunlukla PTC tipi mutasyonlardır ve yanlış anlamlı mutasyonlar da rapor edilmiştir7). Genetik testlerin faydasına ilişkin olarak, izole aniridialı hastaların yaklaşık %85’inde Sanger dizileme veya NGS ile mutasyon saptanır. Ayrıca, MLPA veya CMA ile PAX6 geni içinde veya cis-düzenleyici bölgede delesyonlar yaklaşık %15’inde tespit edilir8).
Wang (2023), yeni bir çerçeve kayması mutasyonu olan c.640_646del (p.R214Pfs*28) tanımlamış ve tam iris yokluğu, foveal hipoplazi, lens ektopisi ve retina dekolmanı olan bir olgu bildirmiştir1).
Ratna ve ark. (2022), Hint bir ailede run-on mutasyonu c.1268A>T (p.*423L) tanımlamıştır. Etkilenen bireylerde tam aniridi, nistagmus, foveal hipoplazi, AAK, lens superior subluksasyonu, yüksek miyopi ve optik atrofi görülmüş olup, C-terminal uzama mutasyonuna bağlı şiddetli bir fenotip sergilenmiştir6).
Sporadik aniridide, PAX6’ya ek olarak WT1 genini de içeren büyük delesyonlar WAGR sendromuna neden olur. WT1 delesyonu varlığında Wilms tümörü riski %50’ye kadar çıkar1). WAGR sendromu şüphesi varsa, genetik testlerle PAX6 ve WT1 delesyonları doğrulanmalı, Wilms tümörü risk değerlendirmesi ve gelişimsel gecikme takibi yapılabilir8). WT1 bölgesinin genetik testlerle değerlendirilmesi zorunludur; sporadik olguların %30’unda 5 yaşına kadar Wilms tümörü geliştiği bildirilmiştir. WT1 geninin PAX6’ya yakın konumlanması nedeniyle, her ikisini de içeren kromozom 11 kısa kol delesyonu (11p13 delesyonu) aniridiye Wilms tümörünün eşlik etmesine yol açar.
Gillespie sendromu, ITPR1 genindeki heterozigot dominant negatif mutasyonlar veya biallelik mutasyonlar sonucu oluşur3). Bugüne kadar moleküler tanısı doğrulanmış 37 olgu bildirilmiş olup, Gly2554 kalıntısı bir sıcak nokta olarak bilinmektedir3).
Aniridi tanı kriterlerine (2020) göre aşağıdaki kriterlerle kesin tanı konur7).
A. Belirtiler
B. Muayene bulguları
C. Ayırıcı tanıda düşünülmesi gereken hastalıklar
E. Genetik testler: PAX6 geninde patojenik mutasyon veya 11p13 bölgesinde delesyon
Tanı kategorileri7):
Nadir hastalık tanısı için şiddet sınıflandırması aşağıdaki 4 aşamada tanımlanmıştır7).
| Şiddet | Tanım |
|---|---|
| Derece I | Tek gözde hastalık, diğer göz sağlıklı |
| Derece II | Her iki gözde hastalık, iyi olan gözde düzeltilmiş görme keskinliği 0.3 veya üzeri |
| Derece III | Her iki gözde hastalık, iyi olan gözde düzeltilmiş görme keskinliği 0.1 veya üzeri ancak 0.3’ten az |
| Derece IV | Her iki gözde hastalık, iyi olan gözde düzeltilmiş görme keskinliği 0.1’den az |
Derece I-III’te bile, glokom gibi nedenlerle görme alanı daralması (Goldmann I/4 hedefi ile merkezi görme alanı 20 derece veya daha az) varsa, bir üst şiddet derecesine geçilir. Derece III ve üzeri, tıbbi maliyet desteği için uygundur 7).
Yarık lamba biyomikroskopisi ile iriste defekt veya hipoplazi görülmesi klinik tanıyı kolaylaştırır. Gonyoskopi veya ultrasonik biyomikroskopi ile kalan iris dokusu değerlendirilir. Ön kamara açısında gelişimsel anormallik olup olmadığı da kontrol edilir.
Aşağıdaki göz komplikasyonları sistematik olarak değerlendirilir:
Aniridinin genetik değerlendirmesinde en önemli hedef, PAX6 delesyonunun WT1 genine kadar uzanıp uzanmadığının doğrulanmasıdır1). Tüm ekzom dizileme veya MLPA yöntemi ile PAX6 ve WT1 bölgelerindeki mutasyon/delesyonlar değerlendirilir1)2).
Sporadik aniridide, WT1 gen delesyonuna bağlı Wilms tümörü riskinin değerlendirilmesi yaşam prognozu ile doğrudan ilişkilidir1). Ailesel olgularda bile fenotipik çeşitlilik nedeniyle genetik test ile kesin tanı ve genetik danışmanlık önerilir.
Aniridi için kesin bir tedavi yoktur. Yönetimin temelini, kalan görmeyi en üst düzeyde kullanmak için az görme rehabilitasyonu ve her bir komplikasyonun bireysel tedavisi oluşturur8).
Korneal stromal opasite için kornea nakli dikkatle değerlendirilmelidir8).
Kornea nakli kısa vadede görme iyileşmesi sağlayabilir ancak maküler hipoplazi gibi eşlik eden durumlar nedeniyle iyileşme sınırlıdır. Uzun vadede glokom ilerlemesi ve greft yetmezliği nedeniyle görme prognozu kötüdür.
Korneal epitelyal kök hücre yetmezliğinde cerrahi tedavi düşünülmelidir 8).
Katarakt cerrahisi, opasite ve fotofobi derecesine göre değerlendirilmelidir 8).
Katarakt, 20 yaşına kadar %50-85 oranında gelişir. Opasite ve fotofobi şiddetine göre cerrahi planlanır. Cerrahi geçiren olguların %66-100’ünde görme iyileşmesi bildirilmiştir, ancak aşağıdaki noktalara dikkat edilmelidir.
Zinn zonülleri zayıf olduğundan, göz içi lens implantasyonu dikkatli endikasyon gerektirir.
Hu ve ark. (2024), şiddetli AAK ile birlikte konjenital aniridisi olan 2 hastada avize retroillüminasyon yardımlı fakoemülsifikasyon uygulamıştır. Kornea bulanıklığı nedeniyle normal intraoperatif görüntüleme zor olsa da, arkadan aydınlatma lens ve ön kapsülün net görüntülenmesini sağlamış ve postoperatif 3. haftada düzeltilmiş görme keskinliği sırasıyla 20/200 ve 20/1000’e iyileşmiştir4).
Glokom görsel prognozu doğrudan etkilediğinden agresif tedavi edilmelidir8).
Glokom başlangıcından sonra aşağıdaki 5 aşamalı algoritmaya göre yönetilir.
İlaç tedavisi: Beta-blokerler, sempatomimetikler ve prostaglandin (PG) analogları etkilidir. Bebeklerde brimonidin (alfa-adrenerjik agonist) santral sinir sistemi depresyonu riski nedeniyle 2 yaş altında kontrendikedir. Kornea epitel hasarı endişesi varsa koruyucu içermeyen formülasyonlar kullanılır.
Drenaj yolu rekonstrüksiyonu (goniotomi/trabekülotomi): İlk cerrahi olarak önerilir16). Profilaktik goniotomi raporları da mevcuttur. Ancak kalan iris trabeküler ağı kaplıyorsa etkisiz olabilir.
Filtrasyon cerrahisi (trabekülektomi): Az sayıda vaka ve orta-kısa dönem raporlarla sınırlıdır. Pediatrik gözlerde sonuçlar genellikle kötüdür ve postoperatif oküler hipotoni yaklaşık %25 oranında görülür13). Postoperatif malign glokom raporları da vardır.
Glokom implant cerrahisi (tüp şant cerrahisi): Baerveldt ve Ahmed tipi cihazlar kullanılabilir. Faki gözlerde tüp yerleşimi kornea merkezine değil teğetsel yönde önerilir. İyi göz içi basınç kontrolü beklenir.
Siliyer cisim fotokoagülasyonu: Son çare. Siliyer cisim kriyoterapisinde çoğu vakanın oküler hipotoniye ilerlediği bildirilmiştir. Siliyer cisim hipoplazisi nedeniyle sağlıklı gözlere göre hipotoni riski daha yüksektir.
Açı gelişimindeki anormallikler nedeniyle, normal açık açılı glokomdan farklı bir yaklaşım gereklidir. İlk seçenek olarak dış akım yolunun yeniden yapılandırılması tercih edilir, ardından tüp şant cerrahisi iyi bir seçenektir. Brimonidin 2 yaş altında kontrendikedir ve antimetabolit kullanımı AAK’yi kötüleştirebileceğinden dikkatli değerlendirme gerektirir8).
Az görme bakımı erken dönemde başlatılmalıdır8).
Temel olarak refraktif düzeltme yapılır; miyopi birliktelik oranı %64’ün üzerindedir.
Fotofobi tedavisi, görsel işlev gelişimi ve yaşam kalitesini korumak için önemlidir8).
Hastaların çoğu normal okullara devam edebilir, ancak büyük puntolu ders kitapları, tablet cihazlar ve kitap sehpaları gibi desteklere ihtiyaç duyarlar. Az görenler sınıfına devam etmek veya körler okulu ile görme engellilere yönelik özel eğitim okulları tarafından sunulan bebek bakımı danışmanlığı ve eğitim danışmanlığı hizmetlerinden yararlanmak da bir seçenektir.
Nisan 2017’den itibaren belirlenmiş nadir hastalık olarak kabul edildiğinden, engelli kimlik kartı alınmamış olsa bile, şiddet derecesi III ve üzeri olan hastalar tıbbi masraf desteği ve yardımcı cihaz desteğinden yararlanabilir7). Desteklenen yardımcı cihazlar arasında düzeltici gözlükler, ışık filtreli gözlükler, kontakt lensler (yapay irisli olanlar dahil), az görenler için gözlükler, görme engelliler için beyaz baston ve protez göz bulunur.
PAX6, 14 ekzon içeren 22 kb’lik bir genomik DNA bölgesine yayılır ve 422 amino asit kodlar1). İki DNA bağlanma alanına (paired domain ve paired-type homeodomain) sahiptir ve C-terminalindeki PST (prolin, serin, treonin açısından zengin) alanı transkripsiyonel aktivatör olarak işlev görür.
PAX6, hücre çoğalmasını, farklılaşmasını, göçünü ve yapışmasını düzenler; hedefleri arasında PAX6’nın kendisinin yanı sıra lens kristalini ve kornea keratinini kodlayan genler bulunur. Yetişkin retinada, lens ve korneada da ifadesi devam eder. PAX6 geni, embriyonik dönemde organ farklılaşmasını yöneten ana kontrol genlerinden biridir.
PAX6 mutasyonlarının çoğu, anlamsız mutasyon aracılı mRNA yıkımı (NMD) yoluyla haplo-yetersizliğe neden olur1). Erken durdurma kodonu (PTC) oluşturan mutasyonlar (anlamsız mutasyonlar, çerçeve kayması mutasyonları ve çoğu splice mutasyonu) tipik aniridi fenotipine yol açar.
Öte yandan, PTC son ekzonda veya sondan bir önceki ekzonun terminal 50 bp’si içinde yer alıyorsa NMD’den kaçar ve kesik protein sentezlenerek şiddetli bir fenotip ortaya çıkabilir1).
PAX6 anlamsız mutasyonu c.282C>A (p.Cys94*) ile trizomi 21’in aynı hastada bir arada görüldüğü nadir bir vaka bildirilmiştir. PAX6 mutasyonu de novo oluşmuş ve tam iki taraflı aniridi, konjenital glokom, AAK ve foveal hipoplazi ile sonuçlanmıştır2).
Kesin bir genotip-fenotip korelasyonu kurulmamış olmakla birlikte, bazı eğilimler bilinmektedir1).
Grant ve Walton’un gonioskopi serilerinde, iris stromasının başlangıçta trabeküler ağ üzerinde öne doğru uzanarak yapışıklık benzeri bir bağlantı oluşturduğu, giderek tabaka haline gelip sonunda açı kapanmasına yol açtığı gösterilmiştir 14). Bu mekanizma glokom gelişiminin ana nedenidir. Patolojik olarak, iris kökünü koruyan düz kas defekti ve açının gelişimsel yetersizliği temel oluşturur.
AAK esas olarak limbal kök hücre yetmezliği (LSCD) tarafından tetiklenir, ancak kornea epitelinin anormal farklılaşması, adezyon bozukluğu, konjonktival hücre infiltrasyonu ve gözyaşı üretiminin yetersizliği de rol oynar. PAX6 tarafından düzenlenen matriks metalloproteinaz 9 (MMP-9) eksikliği, fibrin birikimi ve inflamatuar hücre infiltrasyonuna yol açarak stromadaki kollajen diziliminin bozulmasına ve saydamlığın kaybolmasına neden olur.
AAK 5 evreye ayrılır. Evre I’de sadece periferik epitel anormalliği, Evre II’de santrale ulaşmayan santripetal epitelyal değişiklikler, Evre III’te santral korneada epitelyal değişiklikler ve periferik yüzeyel neovaskülarizasyon, Evre IV’te tüm korneada yüzeyel neovaskülarizasyon, Evre V’te tüm korneada epitelyal anormallik ve derin stromal skar görülür 10).
PAX6 mutasyon durumu ile AAK’nin ilerlemesi arasında ilişki vardır. PTC veya C-terminal uzama mutasyonlarına sahip hastalarda AAK yaşa bağlı olarak ilerlerken, diğer mutasyon tiplerinde ilerleyici olmayan keratopati görülebilir 11).
Gillespie sendromu, ITPR1 genindeki mutasyonlardan kaynaklanır 3). ITPR1, IP3 reseptör ailesinin bir üyesidir ve endoplazmik retikulumda lokalize Ca²⁺ salınım kanalları oluşturur. Dominant negatif mutasyonlar, iris sfinkter kasının oluşumunu ve devamlılığını etkileyerek pupil çevresinde spesifik iris hipoplazisi ve fiks midriyazise yol açar.
Ciaccio ve ark. (2024) Gillespie sendromu literatür incelemesinde, moleküler olarak doğrulanmış 33 vakanın analiziyle motor gelişimin geciktiği ancak zamanla düzeldiği, zihinsel engelin tüm vakalarda görülmediği ve %17’sinde normal zekâ olduğu, nörolojik bulguların ilerleyici olmadığı doğrulanmıştır 3).
Tüm ekzom dizileme teknolojisinin yaygınlaşmasıyla yeni PAX6 mutasyonlarının tanımlanması devam etmektedir. Human PAX6 Mutation Database’de 2018 itibarıyla 491 mutasyon kayıtlı olup, o tarihten bu yana yaklaşık 250 yeni mutasyon daha rapor edilmiştir 1). Kodlamayan bölgelerdeki mutasyonların aniridiye neden olduğu vakalar da tespit edilmekte olup, geleneksel testlerle teşhis edilemeyen olguların aydınlatılması beklenmektedir 9).
Şiddetli AAK’li olgularda katarakt cerrahisi için, avize ters aydınlatma yardımıyla görselleştirme tekniği faydalıdır 4). Bu teknik, Grade 3-4 ileri AAK’li hastalarda bile güvenli fakoemülsifikasyona olanak tanır ve ameliyat sonrası görme iyileşmesi sağlanmıştır.
PAX6 mutasyon tipine göre AAK’nin ilerleme paterninin farklılık gösterdiği giderek daha net anlaşılmaktadır. Genetik test maliyetlerinin düşmesiyle, mutasyon tipine dayalı klinik seyir tahmini ve erken müdahale gerçekçi seçenekler haline gelmektedir.
Aniridi ve trizomi 21’in birlikte görüldüğü olgularda, iki hastalığın bir arada bulunmasına rağmen nispeten hafif bir seyir izlendiği bildirilmiştir 2). Birden fazla genetik bozukluğun aynı hastada bir arada bulunmasının fenotip üzerindeki etkisini anlamak, gelecekteki kişiselleştirilmiş tıp için önemli bilgiler sağlayabilir.
PTC tipi mutasyonlar için bir okuma-bastırma ilacı olan ataluren’in aniridide kullanımı temel araştırma düzeyinde incelenmektedir 8). PAX6 gen tedavisi ile ilgili olarak, Sey mutant fare modelinde AAV-PAX6 vektörü ile gen takviyesine yönelik temel araştırmalar devam etmektedir. Gelecekte klinik çalışmalara geçilmesi beklenmektedir.
iPS hücresi kaynaklı kornea epitel hücre tabakası nakline yönelik klinik çalışmalar yurt içinde ve yurt dışında yürütülmekte olup, AAK için yeni bir tedavi yöntemi olarak dikkat çekmektedir 8). Yapay iris (CustomFlex Artificial Iris vb.) konusunda yurt dışında kullanım deneyimi birikmektedir. Yardımcı araç olarak yapay irisli kontakt lensler sigorta kapsamındadır.
Japonya’da büyük ölçekli kayıt verilerinin birikmesiyle gerçek durumun anlaşılması ve kanıt kalitesinin artırılması gelecekteki önemli konulardır 8). Bireysel gen mutasyonlarına dayalı olarak AAK ilerlemesinin tahmin edilmesi ve erken müdahalenin optimize edilmesi beklenmektedir.