Aniridia đơn độc
Tần suất: Khoảng 2/3 tổng số ca.
Kiểu di truyền: Di truyền trội trên nhiễm sắc thể thường (AD).
Đặc điểm: Do đột biến gen PAX6. Không kèm triệu chứng toàn thân. Tính thâm nhập hoàn toàn nhưng biểu hiện đa dạng.

Bệnh vô mống mắt (Aniridia) là một bệnh bẩm sinh hiếm gặp, đặc trưng bởi sự hình thành không đầy đủ hoặc thiếu hụt mống mắt ở các mức độ khác nhau. Tên gọi “vô mống mắt” là sai lầm, vì khi soi góc tiền phòng hoặc siêu âm sinh hiển vi (UBM) hầu như luôn phát hiện các mảnh mô mống mắt.
Tỷ lệ mắc khoảng 1/40.000 đến 1/100.000, không có sự khác biệt rõ rệt về chủng tộc hay giới tính 1). Trong ICD-10, bệnh được phân loại là Q13.1.
Đây là bệnh lý toàn nhãn cầu, ảnh hưởng không chỉ đến mống mắt mà còn đến giác mạc, thủy tinh thể, góc tiền phòng, hố trung tâm và thần kinh thị giác 1), gây ra nhiều biến chứng mắt đe dọa thị lực. Tiên lượng thị lực nhìn chung kém, thường chỉ đạt thị lực chỉnh kính khoảng 0,1. Phản xạ đồng tử mất nhưng phản xạ điều tiết còn, 60-90% trường hợp là hai mắt.
Ba kiểu hình sau đây đã được ghi nhận.
Aniridia đơn độc
Tần suất: Khoảng 2/3 tổng số ca.
Kiểu di truyền: Di truyền trội trên nhiễm sắc thể thường (AD).
Đặc điểm: Do đột biến gen PAX6. Không kèm triệu chứng toàn thân. Tính thâm nhập hoàn toàn nhưng biểu hiện đa dạng.
Hội chứng WAGR
Tần suất: Một phần trong các ca lẻ tẻ.
Kiểu di truyền: Mất đoạn kề nhau của PAX6 và WT1.
Đặc điểm: Kết hợp u Wilms, bất thường cơ quan sinh dục tiết niệu, chậm phát triển trí tuệ. Nguy cơ khối u lên tới 50%.
Hội chứng Gillespie
Tần suất: Khoảng 2% tổng số ca.
Kiểu di truyền: Đột biến gen ITPR1.
Đặc điểm: Kèm mất điều hòa tiểu não, thiểu năng trí tuệ. Bất thường mống mắt đặc trưng với đồng tử cố định giãn là đặc điểm điển hình3).
Aniridia lẻ tẻ chiếm khoảng 1/3 tổng số ca, do mất đoạn de novo trên 11p13 bao gồm PAX6. Nếu mất đoạn lan đến gen WT1 kề cận sẽ gây hội chứng WAGR1). 25–30% bệnh nhân aniridia lẻ tẻ phát triển u Wilms, với nguy cơ tương đối được báo cáo là 67.
PAX6 là gen kiểm soát chính của sự hình thành mắt, tham gia vào phát triển mắt, ống thần kinh, hành khứu giác, đảo Langerhans tụy và biểu mô khứu giác. Bệnh khởi phát do mất chức năng một alen (haploinsufficiency); nếu cả hai alen bất thường sẽ gây chết phôi thai. Năm 2017, bệnh được chỉ định là bệnh hiếm theo Luật Bệnh hiếm, và từ mức độ nặng III trở lên (xem chi tiết tại mục chẩn đoán/xét nghiệm) sẽ được hỗ trợ chi phí y tế7).
Thể tản phát (đột biến mới) chiếm khoảng 1/3 tổng số ca, có thể phát triển ngay cả khi không có tiền sử gia đình. Ở thể tản phát, có khả năng mắc hội chứng WAGR, do đó xét nghiệm di truyền và siêu âm bụng để tầm soát u Wilms là quan trọng.
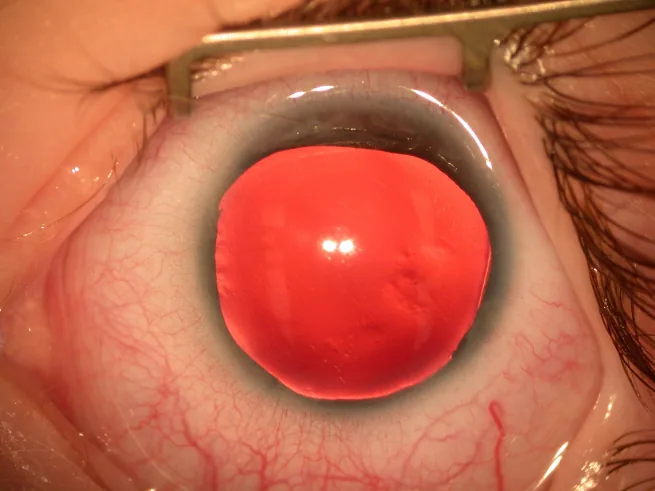
Hầu hết các trường hợp vô mống mắt được phát hiện do bất thường mống mắt và đồng tử khi sinh, hoặc do rung giật nhãn cầu ở giai đoạn trẻ sơ sinh.
Kiểu hình khác nhau giữa các gia đình và trong cùng một gia đình, nhưng sự khác biệt giữa hai mắt thường nhỏ.
Do bất sản hố trung tâm là nguyên nhân chính, thị lực chỉnh kính thường khoảng 0,1-0,2. Tiên lượng thị lực đặc biệt xấu nếu có kèm giảm sản hoàng điểm. Chỉnh tật khúc xạ và chăm sóc thị lực kém từ giai đoạn sơ sinh rất quan trọng cho sự phát triển thị giác.
PAX6 biểu hiện ở mô mắt cũng như hệ thần kinh trung ương, đảo tụy Langerhans và biểu mô khứu giác, do đó có thể gặp các biến chứng ngoài mắt sau 8).
Các yếu tố quan trọng quyết định chức năng thị giác bao gồm glôcôm, bất sản hoàng điểm, rung giật nhãn cầu, bệnh giác mạc, đục thủy tinh thể và bất thường mống mắt. Tổn thương thị trường và thị lực do glôcôm là không hồi phục, do đó quản lý nhãn áp là quan trọng nhất trong theo dõi 8).
Phần lớn các trường hợp vô mống mắt bẩm sinh là do đột biến dị hợp tử ở gen PAX6 nằm trên nhánh ngắn của nhiễm sắc thể 11 (11p13). Cơ chế bệnh sinh chính là do thiếu hụt haplo của PAX6 1).
Gen PAX6 là gen kiểm soát chính trong sự hình thành mắt, đóng vai trò quan trọng trong sự phát triển của mắt, ống thần kinh, khứu cầu và tuyến tụy. Sự phát triển bình thường của mắt cần hai bản sao của PAX6; chỉ mất chức năng một bản sao đã có thể gây ra vô mống mắt 1).
Trong một nghiên cứu đoàn hệ trên bệnh nhân người Trung Quốc, 96,9% trường hợp có đột biến nguyên nhân ở gen PAX6 1). Ở vô mống mắt điển hình, 96% trường hợp phát hiện đột biến gây phân hủy mRNA phụ thuộc mã vô nghĩa (NMD) hoặc mất đoạn lớn 1).
Về mặt bệnh lý, cơ trơn bị thiếu hụt ngoại trừ phần gốc mống mắt, và có sự kém phát triển của góc tiền phòng. Có rối loạn chức năng tế bào gốc biểu mô giác mạc, dẫn đến bất thường biểu mô và màng Bowman, hình thành màng máu (pannus) giàu mạch.
Dưới đây là phân bố các đột biến PAX6 gây ra kiểu hình vô mống mắt.
| Loại đột biến | Tần suất |
|---|---|
| Đột biến vô nghĩa | Khoảng 39% |
| Đột biến dịch khung | Khoảng 25% |
| Đột biến cắt nối | Khoảng 13% |
| Đột biến sai nghĩa | Khoảng 12% |
Đột biến kéo dài (đột biến kéo dài đầu C) chiếm khoảng 5%, xảy ra khi codon kết thúc được chuyển đổi thành codon dịch mã, dẫn đến sản xuất protein PAX6 kéo dài bất thường 6). Đột biến kéo dài đầu C thường đi kèm với thiểu sản mống mắt nặng và suy giảm thị lực nghiêm trọng 1)6).
Đột biến gen chủ yếu là loại PTC, cũng có báo cáo về đột biến sai nghĩa 7). Về hữu ích của xét nghiệm di truyền, Sanger sequencing hoặc NGS phát hiện được gần 85% đột biến ở bệnh nhân isolated aniridia. Ngoài ra, MLPA hoặc CMA phát hiện được gần 15% trường hợp mất đoạn trong gen PAX6 hoặc vùng điều hòa cis 8).
Wang (2023) đã xác định mới đột biến dịch khung c.640_646del (p.R214Pfs*28) và báo cáo một trường hợp biểu hiện thiếu hụt mống mắt hoàn toàn, thiểu sản hố trung tâm, lệch thủy tinh thể và bong võng mạc 1).
Ratna và cộng sự (2022) đã xác định đột biến kéo dài c.1268A>T (p.*423L) trong một gia đình người Ấn Độ. Người bệnh biểu hiện vô mống mắt hoàn toàn, rung giật nhãn cầu, thiểu sản hố trung tâm, AAK, lệch thủy tinh thể lên trên, cận thị nặng và teo thị thần kinh, cho thấy kiểu hình nặng do đột biến kéo dài đầu C 6).
Trong bệnh vô mống mắt lẻ tẻ, mất đoạn lớn bao gồm gen WT1 ngoài PAX6 là nguyên nhân gây hội chứng WAGR. Nguy cơ u Wilms lên tới 50% nếu có mất đoạn WT1 1). Nếu nghi ngờ hội chứng WAGR, xét nghiệm di truyền để xác nhận mất đoạn PAX6 và WT1, từ đó đánh giá nguy cơ u Wilms và theo dõi chậm phát triển 8). Đánh giá vùng WT1 bằng xét nghiệm gen là cần thiết; 30% trường hợp lẻ tẻ phát triển u Wilms trước 5 tuổi. Do gen WT1 nằm gần PAX6, mất đoạn nhánh ngắn nhiễm sắc thể 11 (mất đoạn 11p13) khiến cả hai gen bị mất, gây vô mống mắt kèm u Wilms.
Hội chứng Gillespie xảy ra do đột biến dị hợp tử âm tính trội hoặc đột biến hai alen của gen ITPR1 3). Đến nay, 37 trường hợp đã được xác nhận chẩn đoán phân tử, với gốc Gly2554 được biết đến là điểm nóng 3).
Dựa trên tiêu chuẩn chẩn đoán bệnh vô mống mắt (năm 2020), chẩn đoán xác định dựa trên các tiêu chí sau 7).
A. Triệu chứng
B. Dấu hiệu xét nghiệm
C. Các bệnh cần chẩn đoán phân biệt
E. Xét nghiệm di truyền: Đột biến bệnh lý của gen PAX6 hoặc mất đoạn vùng 11p13
Phân loại chẩn đoán7):
Phân loại mức độ nặng để công nhận bệnh hiếm được quy định theo 4 mức độ sau7).
| Mức độ nặng | Định nghĩa |
|---|---|
| Độ I | Một mắt bị bệnh, mắt kia bình thường |
| Độ II | Cả hai mắt bị bệnh, thị lực chỉnh kính của mắt tốt hơn ≥ 0.3 |
| Độ III | Cả hai mắt bị bệnh, thị lực chỉnh kính của mắt tốt hơn từ 0.1 đến dưới 0.3 |
| Độ IV | Cả hai mắt bị bệnh, thị lực chỉnh kính của mắt tốt hơn dưới 0.1 |
Ngay cả ở độ I–III, nếu kèm theo hẹp thị trường do glôcôm (thị trường trung tâm còn lại trong vòng 20 độ với thị kính Goldmann I/4) thì chuyển lên mức độ nặng hơn một bậc. Mức độ nặng từ độ III trở lên là đối tượng được hỗ trợ chi phí y tế7).
Chẩn đoán lâm sàng dễ dàng khi xác định được thiếu sản hoặc bất sản mống mắt bằng kính hiển vi đèn khe. Đánh giá mô mống mắt còn lại bằng kính hiển vi góc tiền phòng hoặc kính hiển vi siêu âm sinh học. Cũng cần kiểm tra sự hiện diện của bất thường phát triển góc tiền phòng.
Đánh giá một cách có hệ thống các biến chứng mắt sau đây:
Mục tiêu quan trọng nhất trong đánh giá di truyền của bệnh vô mống mắt là xác nhận xem sự mất đoạn PAX6 có mở rộng đến gen WT1 hay không1). Giải trình tự toàn bộ exome hoặc phương pháp MLPA được sử dụng để đánh giá các đột biến/mất đoạn trong vùng PAX6 và WT11)2).
Trong bệnh vô mống mắt lẻ tẻ, việc đánh giá nguy cơ u Wilms do mất đoạn gen WT1 liên quan trực tiếp đến tiên lượng sống1). Ngay cả trong trường hợp gia đình, do tính đa dạng về kiểu hình, xét nghiệm di truyền để chẩn đoán xác định và tư vấn di truyền được khuyến cáo.
Không có phương pháp điều trị triệt để cho bệnh vô mống mắt. Trọng tâm quản lý là chăm sóc thị lực kém để tận dụng tối đa thị lực còn lại và điều trị riêng lẻ từng biến chứng8).
Cần thận trọng khi quyết định ghép giác mạc cho tình trạng đục nhu mô giác mạc8).
Ghép giác mạc có thể cải thiện thị lực trong ngắn hạn, nhưng sự cải thiện bị hạn chế do các bệnh đi kèm như thiểu sản hoàng điểm. Về lâu dài, tiến triển của bệnh tăng nhãn áp và suy chức năng mảnh ghép dẫn đến tiên lượng thị lực xấu.
Trong bệnh suy kiệt tế bào gốc biểu mô giác mạc, cần xem xét điều trị phẫu thuật 8).
Phẫu thuật đục thủy tinh thể được xem xét dựa trên mức độ đục và chói sáng 8).
Đục thủy tinh thể xảy ra ở 50–85% bệnh nhân trước 20 tuổi. Phẫu thuật được lên kế hoạch dựa trên mức độ đục và chói sáng. Có báo cáo cho thấy 66–100% ca phẫu thuật cải thiện thị lực, nhưng cần lưu ý các điểm sau:
Do dây chằng Zinn yếu, việc đặt thủy tinh thể nhân tạo cần được chỉ định thận trọng.
Hu và cộng sự (2024) đã thực hiện phẫu thuật hút thủy tinh thể bằng siêu âm có hỗ trợ chiếu sáng ngược dạng đèn chùm trên 2 bệnh nhân mắc chứng vô mống mắt bẩm sinh kèm AAK nặng. Do độ mờ giác mạc gây khó khăn cho việc quan sát trong phẫu thuật thông thường, nhưng nhờ ánh sáng từ phía sau, thủy tinh thể và bao trước có thể được quan sát rõ ràng, và thị lực chỉnh kính đã cải thiện lên lần lượt 20/200 và 20/1000 sau 3 tuần phẫu thuật4).
Bệnh tăng nhãn áp cần được điều trị tích cực vì ảnh hưởng trực tiếp đến tiên lượng thị lực8).
Sau khi khởi phát bệnh tăng nhãn áp, việc quản lý được thực hiện dựa trên thuật toán 5 bước sau đây.
Điều trị bằng thuốc: Thuốc chẹn beta, thuốc kích thích giao cảm, thuốc liên quan đến prostaglandin (PG) có hiệu quả. Brimonidine (thuốc kích thích thụ thể alpha-adrenergic) ở trẻ nhỏ có nguy cơ ức chế thần kinh trung ương, do đó chống chỉ định cho trẻ dưới 2 tuổi. Nếu có nguy cơ tổn thương biểu mô giác mạc, sử dụng chế phẩm không chứa chất bảo quản.
Phẫu thuật tái tạo đường dẫn lưu (phẫu thuật mở góc tiền phòng/phẫu thuật mở bè củng giác mạc): Được khuyến cáo như phẫu thuật đầu tay16). Cũng có báo cáo về phẫu thuật mở góc tiền phòng dự phòng. Tuy nhiên, có thể không hiệu quả trong các trường hợp mống mắt còn lại che phủ bè củng giác mạc.
Phẫu thuật lọc (cắt bè củng giác mạc): Chỉ có báo cáo về một số ít trường hợp và theo dõi trung hạn. Xu hướng kết quả kém ở mắt trẻ em, lỗ rò nhãn cầu sau phẫu thuật xảy ra ở khoảng 25%13). Cũng có báo cáo về glôcôm ác tính sau phẫu thuật.
Phẫu thuật cấy ghép glôcôm (phẫu thuật ống shunt): Có thể sử dụng thiết bị loại Baerveldt và Ahmed. Khuyến cáo đặt ống vào mắt còn thể thủy tinh theo hướng tiếp tuyến thay vì hướng vào trung tâm giác mạc. Có thể kiểm soát nhãn áp tốt.
Phẫu thuật đông tụ thể mi: biện pháp cuối cùng. Có báo cáo cho thấy nhiều trường hợp đông lạnh thể mi dẫn đến teo nhãn cầu. Do thể mi kém phát triển, nguy cơ teo nhãn cầu cao hơn so với mắt khỏe mạnh.
Do có bất thường phát triển ở góc tiền phòng, cần có cách tiếp cận khác với bệnh tăng nhãn áp góc mở thông thường. Lựa chọn đầu tiên là phẫu thuật tái tạo đường dẫn lưu, sau đó phẫu thuật đặt ống dẫn lưu là lựa chọn tốt. Brimonidine chống chỉ định ở trẻ dưới 2 tuổi, và việc sử dụng thuốc chống chuyển hóa có thể làm nặng thêm AAK, do đó cần cân nhắc thận trọng 8).
Chăm sóc thị lực kém nên được bắt đầu sớm8).
Chỉnh khúc xạ là cơ bản, tỷ lệ kèm cận thị được báo cáo trên 64%.
Điều trị chứng sợ ánh sáng rất quan trọng để duy trì sự phát triển chức năng thị giác và chất lượng cuộc sống8).
Hầu hết bệnh nhân có thể học tại lớp học phổ thông, nhưng cần hỗ trợ như sách giáo khoa phóng to, máy tính bảng, giá đọc sách. Các lựa chọn khác bao gồm học tại lớp dành cho trẻ khiếm thị, hoặc sử dụng dịch vụ tư vấn nuôi dạy trẻ và tư vấn giáo dục từ trường mù hoặc trường hỗ trợ đặc biệt về thị giác.
Kể từ tháng 4 năm 2017, bệnh này được công nhận là bệnh hiếm được chỉ định, do đó ngay cả khi chưa có giấy chứng nhận khuyết tật thể chất, nếu mức độ nghiêm trọng từ độ III trở lên, bệnh nhân sẽ được hỗ trợ chi phí y tế và cấp dụng cụ chỉnh hình7). Các dụng cụ chỉnh hình bao gồm kính điều chỉnh, kính chống chói, kính áp tròng (bao gồm loại có mống mắt nhân tạo), kính cho người khiếm thị, gậy an toàn cho người khiếm thị và mắt giả.
PAX6 trải dài trên 22kb DNA bộ gen bao gồm 14 exon, mã hóa 422 axit amin1). Nó có hai vùng liên kết DNA (vùng ghép đôi và vùng homeodomain kiểu ghép đôi), và vùng PST (giàu proline, serine, threonine) ở đầu C hoạt động như một yếu tố kích hoạt phiên mã.
PAX6 kiểm soát sự tăng sinh, biệt hóa, di chuyển và kết dính của tế bào; các mục tiêu của nó bao gồm chính PAX6, cũng như các gen mã hóa crystallin thủy tinh thể và keratin giác mạc. Sự biểu hiện vẫn tiếp tục ở võng mạc, thủy tinh thể và giác mạc của người trưởng thành. Gen PAX6 là một trong những gen kiểm soát chính điều phối sự biệt hóa cơ quan trong giai đoạn phôi thai.
Hầu hết các đột biến PAX6 gây ra thiếu hụt đơn bội thông qua phân hủy mRNA phụ thuộc đột biến vô nghĩa (NMD)1). Các đột biến tạo ra codon kết thúc sớm (PTC) (đột biến vô nghĩa, đột biến dịch khung, hầu hết đột biến ghép nối) dẫn đến kiểu hình aniridia điển hình.
Ngược lại, nếu PTC nằm ở exon cuối cùng hoặc trong vòng 50bp cuối của exon áp chót, nó có thể thoát khỏi NMD, dẫn đến dịch mã protein bị cắt ngắn và có thể gây ra kiểu hình nghiêm trọng1).
Một trường hợp hiếm gặp đã được báo cáo về đột biến vô nghĩa PAX6 c.282C>A (p.Cys94*) kết hợp với trisomy 21 ở cùng một bệnh nhân. Đột biến PAX6 xảy ra de novo, gây ra aniridia hoàn toàn hai mắt, glôcôm bẩm sinh, AAK và bất sản hố trung tâm2).
Mặc dù chưa thiết lập được mối tương quan kiểu gen – kiểu hình rõ ràng, một số xu hướng đã được biết đến1).
Trong loạt nghiên cứu soi góc tiền phòng của Grant và Walton, ban đầu nhu mô mống mắt kéo dài về phía trước trên bè củng giác mạc, tạo thành sự bám dính giống như dính, dần dần hình thành dạng tấm và cuối cùng dẫn đến tắc góc tiền phòng14). Cơ chế này là yếu tố chính gây ra bệnh tăng nhãn áp. Về mặt bệnh lý, sự thiếu hụt cơ trơn ngoại trừ chân mống mắt và sự phát triển bất thường của góc tiền phòng là nền tảng.
AAK chủ yếu do thiếu hụt tế bào gốc vùng rìa (LSCD), nhưng cũng liên quan đến biệt hóa bất thường biểu mô giác mạc, dính bất thường, xâm nhập tế bào kết mạc và thiếu sản xuất nước mắt. Sự thiếu hụt matrix metalloproteinase-9 (MMP-9) do PAX6 điều hòa gây tích tụ fibrin và xâm nhập tế bào viêm, dẫn đến rối loạn sắp xếp collagen nhu mô và mất độ trong suốt.
AAK được phân loại thành 5 giai đoạn. Giai đoạn I chỉ bất thường biểu mô ngoại vi, giai đoạn II thay đổi biểu mô hướng tâm (chưa đến trung tâm), giai đoạn III thay đổi biểu mô giác mạc trung tâm và tân mạch nông ngoại vi, giai đoạn IV tân mạch nông toàn bộ giác mạc, giai đoạn V bất thường biểu mô toàn bộ giác mạc và sẹo nhu mô sâu10).
Có mối liên quan giữa tình trạng đột biến PAX6 và tiến triển của AAK. Ở bệnh nhân có đột biến PTC hoặc kéo dài đầu C, AAK tiến triển phụ thuộc vào tuổi, trong khi các dạng đột biến khác có thể gây bệnh giác mạc không tiến triển11).
Hội chứng Gillespie do đột biến gen ITPR1 gây ra3). ITPR1 là thành viên của họ thụ thể IP3, tạo thành kênh giải phóng Ca²⁺ và khu trú ở lưới nội chất. Đột biến âm tính trội ảnh hưởng đến sự hình thành và duy trì cơ thắt mống mắt, dẫn đến thiểu sản mống mắt đặc hiệu quanh đồng tử và giãn đồng tử cố định.
Trong tổng quan y văn về hội chứng Gillespie của Ciaccio và cộng sự (2024), phân tích 33 trường hợp được xác nhận phân tử cho thấy phát triển vận động chậm nhưng cải thiện theo thời gian, không phải tất cả đều có khuyết tật trí tuệ (17% có trí tuệ bình thường), và các dấu hiệu thần kinh không tiến triển3).
Nhờ sự phổ biến của kỹ thuật giải trình tự toàn bộ exome, các đột biến PAX6 mới liên tục được xác định. Tính đến năm 2018, Cơ sở dữ liệu đột biến PAX6 ở người đã ghi nhận 491 đột biến, và kể từ đó, khoảng 250 đột biến mới đã được báo cáo 1). Các đột biến ở vùng không mã hóa cũng đang được xác định là nguyên nhân gây bệnh vô mống mắt, hứa hẹn làm sáng tỏ các trường hợp không được chẩn đoán bằng xét nghiệm thông thường 9).
Đối với phẫu thuật đục thủy tinh thể ở các trường hợp AAK nặng, kỹ thuật chiếu sáng nội soi hỗ trợ bằng đèn chandelier rất hữu ích 4). Kỹ thuật này cho phép thực hiện phacoemulsification an toàn ngay cả ở bệnh nhân có AAK độ 3-4, và cải thiện thị lực sau phẫu thuật.
Ngày càng rõ ràng rằng các loại đột biến PAX6 khác nhau dẫn đến các mô hình tiến triển AAK khác nhau. Với chi phí xét nghiệm di truyền giảm, việc dự đoán diễn tiến lâm sàng dựa trên loại đột biến và can thiệp sớm đang trở thành lựa chọn khả thi.
Một trường hợp bệnh vô mống mắt kết hợp với hội chứng Down (trisomy 21) đã được báo cáo có diễn tiến tương đối nhẹ mặc dù có sự hiện diện của cả hai bệnh 2). Hiểu được ảnh hưởng của nhiều rối loạn di truyền cùng tồn tại ở một bệnh nhân lên kiểu hình có thể mang lại những hiểu biết quan trọng cho y học cá thể hóa trong tương lai.
Việc ứng dụng thuốc đọc qua (ataluren) cho các đột biến loại PTC trong bệnh vô mống mắt đang được nghiên cứu ở cấp độ cơ bản 8). Về liệu pháp gen PAX6, các nghiên cứu cơ bản về bổ sung gen bằng vector AAV-PAX6 trên mô hình chuột đột biến Sey đang được tiến hành. Kỳ vọng sẽ có các thử nghiệm lâm sàng trong tương lai.
Các thử nghiệm ghép tấm tế bào biểu mô giác mạc có nguồn gốc từ tế bào iPS đang được tiến hành trong và ngoài nước, thu hút sự chú ý như một phương pháp điều trị mới cho AAK 8). Mống mắt nhân tạo (như CustomFlex Artificial Iris) đã tích lũy kinh nghiệm sử dụng ở nước ngoài. Kính áp tròng có mống mắt nhân tạo như một dụng cụ hỗ trợ được bảo hiểm chi trả.
Việc tích lũy dữ liệu đăng ký quy mô lớn tại Nhật Bản để nắm bắt thực trạng và nâng cao chất lượng bằng chứng là những nhiệm vụ quan trọng trong tương lai 8). Kỳ vọng sẽ tối ưu hóa dự đoán tiến triển AAK dựa trên từng đột biến gen và can thiệp sớm.