Isolierte Aniridie
Häufigkeit: etwa 2/3 aller Fälle.
Vererbungsmodus: autosomal-dominant (AD).
Merkmale: Verursacht durch PAX6-Genmutation. Keine systemischen Symptome. Penetranz vollständig, Expressivität variabel.

Aniridie ist eine seltene angeborene Erkrankung, die durch eine unterschiedlich starke Hypoplasie oder einen Defekt der Iris gekennzeichnet ist. Der Begriff „Aniridie“ ist irreführend, da bei der Gonioskopie oder Ultraschallbiomikroskopie (UBM) fast immer Irisfragmente nachweisbar sind.
Die Prävalenz wird auf etwa 1/40.000 bis 1/100.000 geschätzt; es gibt keine signifikanten Unterschiede bezüglich Ethnie oder Geschlecht 1). In der ICD-10 wird sie unter Q13.1 klassifiziert.
Aniridie ist eine panokuläre Erkrankung, die nicht nur die Iris, sondern auch Hornhaut, Linse, Kammerwinkel, Fovea und Sehnerv betrifft 1) und zu verschiedenen das Sehvermögen bedrohenden Augenkomplikationen führt. Die Sehprognose ist meist schlecht; die korrigierte Sehschärfe bleibt oft bei etwa 0,1. Die Pupillenreaktion fehlt, aber die Akkommodation ist erhalten; 60–90 % der Fälle sind bilateral.
Die folgenden drei Phänotypen werden unterschieden.
Isolierte Aniridie
Häufigkeit: etwa 2/3 aller Fälle.
Vererbungsmodus: autosomal-dominant (AD).
Merkmale: Verursacht durch PAX6-Genmutation. Keine systemischen Symptome. Penetranz vollständig, Expressivität variabel.
WAGR-Syndrom
Häufigkeit: ein Teil der sporadischen Fälle.
Vererbungsmodus: benachbarte Deletion von PAX6 und WT1.
Merkmale: Assoziiert mit Wilms-Tumor, Urogenitalanomalien und geistiger Behinderung. Tumorrisiko bis zu 50%.
Gillespie-Syndrom
Häufigkeit: etwa 2% aller Fälle.
Vererbungsmodus: ITPR1-Genmutation.
Merkmale: Begleitet von zerebellärer Ataxie und geistiger Behinderung. Charakteristische Irisanomalie mit fixierter Mydriasis 3).
Sporadische Aniridie macht etwa 1/3 aller Fälle aus und wird durch eine de novo-Deletion von 11p13 einschließlich PAX6 verursacht. Wenn die Deletion auf das benachbarte WT1-Gen übergreift, führt dies zum WAGR-Syndrom 1). 25–30% der sporadischen Aniridie-Patienten entwickeln einen Wilms-Tumor, das relative Risiko wird mit 67 angegeben.
PAX6 ist das Master-Kontrollgen der Augenentwicklung und an der Entwicklung von Auge, Neuralrohr, Riechkolben, Langerhans-Inseln der Bauchspeicheldrüse und Riechepithel beteiligt. Die Erkrankung entsteht durch Haploinsuffizienz (Funktionsverlust eines Allels); bei beidseitiger Anomalie ist die Embryonalentwicklung letal. Seit 2017 ist die Aniridie als seltene Erkrankung nach dem japanischen Gesetz über seltene Erkrankungen eingestuft; ab Schweregrad III (siehe Abschnitt Diagnose/Untersuchung) besteht Anspruch auf Kostenübernahme 7).
Sporadische Fälle (Neumutationen) machen etwa ein Drittel aller Fälle aus und treten auch ohne Familienanamnese auf. Bei sporadischen Fällen besteht die Möglichkeit eines WAGR-Syndroms, daher sind Gentests und abdominale Ultraschalluntersuchungen zum Wilms-Tumor-Screening wichtig.
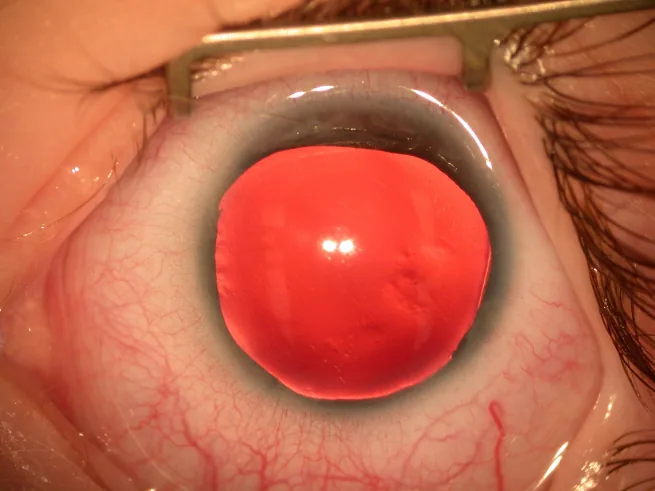
Die meisten Aniridie-Fälle werden durch eine Iris- und Pupillenanomalie bei der Geburt oder durch Nystagmus im Säuglingsalter entdeckt.
Der Phänotyp variiert zwischen und innerhalb von Familien, die Unterschiede zwischen linkem und rechtem Auge sind jedoch meist gering.
Aufgrund der Foveahypoplasie liegt die korrigierte Sehschärfe meist bei etwa 0,1–0,2. Bei gleichzeitiger Makulahypoplasie ist die Sehprognose besonders schlecht. Eine Refraktionskorrektur und Sehbehinderungshilfe ab dem Säuglingsalter sind für die visuelle Entwicklung wichtig.
Da PAX6 neben dem Augengewebe auch im Zentralnervensystem, in den Langerhans-Inseln der Bauchspeicheldrüse und im Riechepithel exprimiert wird, können folgende extraokuläre Komplikationen auftreten 8).
Die wichtigsten Faktoren für die Sehfunktion sind Glaukom, Makulahypoplasie, Nystagmus, Hornhauterkrankung, Katarakt und Irisdysplasie. Da die durch Glaukom verursachten Gesichtsfeld- und Sehschärfeverluste irreversibel sind, ist das Augeninnendruckmanagement bei der Nachsorge von größter Bedeutung.8)
Die meisten Fälle von angeborener Aniridie werden durch heterozygote Mutationen im PAX6-Gen auf dem kurzen Arm von Chromosom 11 (11p13) verursacht. Der Hauptmechanismus ist Haploinsuffizienz von PAX6.1)
Das PAX6-Gen ist ein Master-Kontrollgen für die Augenentwicklung und spielt eine wichtige Rolle bei der Entwicklung von Augen, Neuralrohr, Riechkolben und Bauchspeicheldrüse. Für eine normale Augenentwicklung sind zwei Kopien von PAX6 erforderlich; der Verlust einer Kopie führt bereits zur Aniridie.1)
In einer Kohortenstudie mit chinesischen Patienten wurde bei 96,9% der Fälle eine ursächliche Mutation im PAX6-Gen identifiziert.1) Bei typischer Aniridie werden in 96% der Fälle Mutationen gefunden, die einen nonsense-vermittelten mRNA-Abbau (NMD) induzieren, oder große Deletionen.1)
Pathologisch zeigt sich ein Fehlen der glatten Muskulatur bei erhaltenem Iriswurzelrest sowie eine Fehlentwicklung des Kammerwinkels. Es liegt eine Funktionsstörung der limbalen Stammzellen vor, die zu Anomalien des Epithels und der Bowman-Membran sowie zur Bildung eines vaskularisierten Pannus führt.
Die Verteilung der PAX6-Mutationen, die den Phänotyp der Aniridie verursachen, ist unten dargestellt.
| Mutationstyp | Häufigkeit |
|---|---|
| Nonsense-Mutation | ca. 39% |
| Frame-Shift-Mutation | ca. 25% |
| Spleißmutation | ca. 13% |
| Missense-Mutation | etwa 12% |
Readthrough-Mutationen (C-terminale Verlängerungsmutationen) machen etwa 5% aus, wobei durch die Umwandlung eines Stopp-Codons in ein Translationscodon ein abnormal verlängertes PAX6-Protein produziert wird 6). C-terminale Verlängerungsmutationen gehen häufig mit schwerer Iris-Hypoplasie und hochgradiger Sehbehinderung einher 1)6).
Genmutationen sind häufig vom PTC-Typ, es gibt aber auch Berichte über Missense-Mutationen 7). Zur Nützlichkeit genetischer Tests: Mittels Sanger-Sequenzierung oder NGS werden bei fast 85% der isolierten Aniridie Mutationen nachgewiesen. Zusätzlich werden bei fast 15% Deletionen innerhalb des PAX6-Gens oder der cis-regulatorischen Region mittels MLPA oder CMA gefunden 8).
Wang (2023) identifizierte eine neue Frameshift-Mutation c.640_646del (p.R214Pfs*28) und berichtete über einen Fall mit vollständigem Iris-Kolobom, Fovea-Hypoplasie, Linsenluxation und Netzhautablösung 1).
Ratna et al. (2022) identifizierten in einer indischen Familie eine Readthrough-Mutation c.1268A>T (p.*423L). Die Betroffenen zeigten eine komplette Aniridie, Nystagmus, Fovea-Hypoplasie, AAK, superiore Linsensubluxation, hohe Myopie und Optikusatrophie, was auf einen schweren Phänotyp durch C-terminale Verlängerungsmutation hinweist 6).
Bei sporadischer Aniridie verursachen große Deletionen, die neben PAX6 auch das WT1-Gen umfassen, das WAGR-Syndrom. Bei Vorliegen einer WT1-Deletion beträgt das Wilms-Tumor-Risiko bis zu 50% 1). Bei Verdacht auf WAGR-Syndrom können genetische Tests Deletionen von PAX6 und WT1 bestätigen, was eine Risikobewertung für Wilms-Tumor und eine Nachbeobachtung auf Entwicklungsverzögerung ermöglicht 8). Die Bewertung der WT1-Region durch Gentests ist unerlässlich; bei 30% der sporadischen Fälle tritt ein Wilms-Tumor vor dem 5. Lebensjahr auf. Da das WT1-Gen in der Nähe von PAX6 liegt, führt eine Deletion des kurzen Arms von Chromosom 11 (11p13-Deletion), die beide Gene betrifft, zu einer Aniridie mit Wilms-Tumor.
Das Gillespie-Syndrom wird durch heterozygote dominant-negative Mutationen oder biallelische Mutationen im ITPR1-Gen verursacht 3). Bisher wurden 37 Fälle mit molekularer Diagnose berichtet, wobei der Gly2554-Rest als Hotspot bekannt ist 3).
Basierend auf den Diagnosekriterien für Aniridie (2020) wird die Diagnose anhand der folgenden Kriterien gestellt7).
A. Symptome
B. Untersuchungsbefunde
C. Differentialdiagnosen
E. Genetische Tests: Pathogene Mutation im PAX6-Gen oder Deletion der Region 11p13
Diagnosekategorien7):
Die Schweregradeinteilung für die Anerkennung als seltene Erkrankung ist in vier Stufen festgelegt7).
| Schweregrad | Definition |
|---|---|
| Grad I | Ein Auge betroffen, das andere gesund |
| Grad II | Beidseitige Erkrankung, korrigierter Visus des besseren Auges ≥ 0,3 |
| Grad III | Beidseitige Erkrankung, korrigierter Visus des besseren Auges ≥ 0,1 und < 0,3 |
| Grad IV | Beidseitige Erkrankung, korrigierter Visus des besseren Auges < 0,1 |
Auch bei Grad I–III erfolgt bei Vorliegen einer Gesichtsfeldeinschränkung (zentrales Restgesichtsfeld ≤ 20° mit Goldmann I/4-Marke) durch Glaukom etc. eine Einstufung in den nächsthöheren Schweregrad. Ab Grad III besteht Anspruch auf Kostenübernahme durch die medizinische Versorgung 7).
Die klinische Diagnose ist einfach, wenn mittels Spaltlampenmikroskopie ein Irisdefekt oder eine Irisdysplasie festgestellt wird. Die Beurteilung des verbleibenden Irisgewebes erfolgt mittels Gonioskopie oder Ultraschallbiomikroskopie. Auch das Vorliegen von Entwicklungsanomalien des Kammerwinkels wird überprüft.
Die folgenden Augenkomplikationen werden systematisch beurteilt:
Das wichtigste Ziel der genetischen Beurteilung bei Aniridie ist die Bestätigung, ob die PAX6-Deletion bis zum WT1-Gen reicht 1). Mittels Exom-Sequenzierung oder MLPA werden Mutationen und Deletionen in den PAX6- und WT1-Regionen bewertet 1)2).
Bei sporadischer Aniridie ist die Bewertung des Wilms-Tumor-Risikos durch eine WT1-Gendeletion direkt mit der Lebensprognose verbunden 1). Auch bei familiären Fällen wird aufgrund der phänotypischen Variabilität eine genetische Bestätigungsdiagnostik und genetische Beratung empfohlen.
Es gibt keine kausale Therapie für Aniridie. Die Behandlung konzentriert sich auf Low-Vision-Versorgung zur bestmöglichen Nutzung des Restsehvermögens und die individuelle Behandlung der jeweiligen Komplikationen 8).
Eine Hornhauttransplantation bei Hornhauttrübung ist zurückhaltend zu indizieren 8).
Eine Hornhauttransplantation kann kurzfristig eine Sehverbesserung bewirken, jedoch ist die Verbesserung aufgrund von Begleiterkrankungen wie Makulahypoplasie begrenzt. Langfristig ist die Sehprognose durch das Fortschreiten des Glaukoms und Transplantatversagen ungünstig.
Bei Hornhautepithelstammzellinsuffizienz sollte eine operative Behandlung in Betracht gezogen werden 8).
Eine Kataraktoperation sollte in Abhängigkeit vom Ausmaß der Trübung und Photophobie erwogen werden 8).
Katarakt tritt bei 50–85 % der Patienten bis zum 20. Lebensjahr auf. Die Operation wird je nach Stärke der Trübung und Photophobie geplant. Es wird berichtet, dass bei 66–100 % der operierten Fälle eine Verbesserung der Sehschärfe erreicht wird, jedoch sind folgende Punkte zu beachten:
Aufgrund der Fragilität der Zonula Zinnii ist die Implantation einer Intraokularlinse nur mit Vorsicht indiziert.
Hu et al. (2024) führten bei zwei Fällen von angeborener Aniridie mit schwerer AAK eine Phakoemulsifikation unter Zuhilfenahme einer Kronleuchter-Gegenbeleuchtung durch. Trotz erschwerter intraoperativer Visualisierung durch Hornhauttrübung ermöglichte die Beleuchtung von hinten eine klare Darstellung von Linse und Vorderkapsel, sodass sich der korrigierte Visus 3 Wochen postoperativ auf 20/200 bzw. 20/1000 verbesserte 4).
Das Glaukom wird aktiv behandelt, da es direkten Einfluss auf die visuelle Prognose hat 8).
Nach Auftreten eines Glaukoms erfolgt die Behandlung nach folgendem 5-Stufen-Algorithmus.
Medikamentöse Therapie: Betablocker, Sympathomimetika und Prostaglandin (PG)-Analoga sind wirksam. Brimonidin (α-Adrenozeptor-Agonist) ist bei Säuglingen aufgrund des Risikos einer ZNS-Depression unter 2 Jahren kontraindiziert. Bei Hornhautepithelschäden sollten konservierungsmittelfreie Präparate verwendet werden.
Abflusswegsrekonstruktion (Goniotomie, Trabekulotomie): Als Ersteingriff empfohlen 16). Es gibt auch Berichte über prophylaktische Goniotomie. Bei Fällen, bei denen Restiris das Trabekelwerk bedeckt, kann dies jedoch unwirksam sein.
Fistulierende Operation (Trabekulektomie): Nur wenige Fallserien mit kurzer bis mittlerer Nachbeobachtungszeit. Bei kindlichen Augen tendenziell schlechtere Ergebnisse, postoperative Bulbushypotonie in etwa 25 % 13). Auch über postoperatives malignes Glaukom wurde berichtet.
Glaukom-Implantat-Operation (Tube-Shunt-Operation): Baerveldt- und Ahmed-Implantate sind verfügbar. Bei phaken Augen wird die Tubusinsertion tangential statt zur Hornhautmitte hin empfohlen. Gute Augeninnendruckkontrolle ist zu erwarten.
Zyklophotokoagulation: Ultima Ratio. Bei Kryokoagulation des Ziliarkörpers wurde in vielen Fällen eine Bulbushypotonie berichtet. Aufgrund der Ziliarkörperhypoplasie ist das Risiko einer Hypotonie höher als bei gesunden Augen.
Aufgrund der zugrunde liegenden Entwicklungsanomalie des Kammerwinkels ist ein anderer Ansatz als beim primären Offenwinkelglaukom erforderlich. Zunächst wird eine Trabekulotomie durchgeführt, und anschließend ist eine Tube-Shunt-Operation eine gute Option. Brimonidin ist bei Kindern unter 2 Jahren kontraindiziert, und der Einsatz von Antimetaboliten kann AAK verschlechtern, daher ist eine sorgfältige Abwägung erforderlich 8).
Die Sehbehindertenversorgung sollte frühzeitig eingeleitet werden 8).
Die Refraktionskorrektur ist grundlegend; die Komorbiditätsrate von Myopie beträgt über 64 %.
Die Behandlung der Photophobie ist wichtig, um die Sehentwicklung und die Lebensqualität zu erhalten 8).
Die meisten Patienten können eine Regelschule besuchen, benötigen jedoch Hilfsmittel wie vergrößerte Schulbücher, Tablets und Buchstützen. Auch der Besuch einer Sehbehindertenklasse oder die Inanspruchnahme von Erziehungs- und Bildungsberatung durch Blindenschulen oder spezielle Sehförderschulen sind Optionen.
Seit April 2017 ist die Erkrankung als designierte seltene Krankheit anerkannt. Daher haben Patienten, auch ohne Schwerbehindertenausweis, ab Schweregrad III Anspruch auf Kostenübernahme für medizinische Behandlungen und Hilfsmittel7). Zu den förderfähigen Hilfsmitteln gehören Korrekturbrillen, Sonnenschutzbrillen, Kontaktlinsen (einschließlich solcher mit künstlicher Iris), Sehbehindertenbrillen, weiße Langstöcke für Sehbehinderte und Augenprothesen.
PAX6 erstreckt sich über 22 kb genomischer DNA mit 14 Exons und kodiert 422 Aminosäuren1). Es besitzt zwei DNA-Bindungsdomänen (gepaarte Domäne und gepaarte Homöodomäne), und die C-terminale PST-Domäne (reich an Prolin, Serin und Threonin) fungiert als Transkriptionsaktivator.
PAX6 reguliert Zellproliferation, -differenzierung, -migration und -adhäsion. Zu seinen Zielgenen gehören neben PAX6 selbst Gene, die für Linsen-Crystallin und Hornhaut-Keratin kodieren. Die Expression bleibt in der Netzhaut, Linse und Hornhaut von Erwachsenen bestehen. PAX6 ist eines der Master-Kontrollgene, das die Organentwicklung in der Embryonalphase steuert.
Die meisten PAX6-Mutationen verursachen über den Nonsense-vermittelten mRNA-Abbau (NMD) eine Haploinsuffizienz1). Mutationen, die ein vorzeitiges Stoppcodon (PTC) einführen (Nonsense-Mutationen, Frameshift-Mutationen und die meisten Spleißmutationen), führen zum typischen Phänotyp der Aniridie.
Liegt das PTC hingegen im letzten Exon oder innerhalb der letzten 50 bp des vorletzten Exons, kann es dem NMD entgehen, und ein verkürztes Protein wird translatiert, was zu einem schweren Phänotyp führen kann1).
Es wurde ein seltener Fall berichtet, bei dem eine PAX6-Nonsense-Mutation c.282C>A (p.Cys94*) und eine Trisomie 21 bei demselben Patienten auftraten. Die PAX6-Mutation trat de novo auf und führte zu vollständiger bilateraler Aniridie, angeborenem Glaukom, AAK und Foveahypoplasie2).
Obwohl keine eindeutige Genotyp-Phänotyp-Korrelation etabliert ist, sind einige Tendenzen bekannt1).
In der gonioskopischen Serie von Grant und Walton zeigte sich, dass das Irisstroma zunächst nach anterior auf das Trabekelwerk vorwächst und adhäsionsartige Anheftungen bildet, die sich allmählich zu einer Schicht ausdehnen und schließlich zum Kammerwinkelverschluss führen 14). Dieser Mechanismus ist der Hauptfaktor für die Glaukomentstehung. Pathologisch liegen ein Defekt der glatten Muskulatur unter Erhalt der Iriswurzel und eine Kammerwinkeldysgenesie zugrunde.
AAK wird hauptsächlich durch einen Limbusstammzellmangel (LSCD) verursacht, aber auch abnorme Differenzierung des Hornhautepithels, Adhäsionsstörungen, Infiltration von Bindehautzellen und Tränenproduktionsmangel spielen eine Rolle. Ein Mangel an Matrix-Metalloproteinase 9 (MMP-9), die durch PAX6 reguliert wird, führt zu Fibrinablagerung und Infiltration von Entzündungszellen, wodurch die Kollagenanordnung im Stroma gestört wird und die Transparenz verloren geht.
AAK wird in 5 Stadien eingeteilt. Stadium I zeigt nur periphere Epithelanomalien, Stadium II zentripetale Epithelveränderungen (Zentrum noch nicht erreicht), Stadium III Epithelveränderungen der zentralen Hornhaut und oberflächliche Neovaskularisation der Peripherie, Stadium IV oberflächliche Neovaskularisation der gesamten Hornhaut, Stadium V Epithelanomalien der gesamten Hornhaut und tiefe Stromanarben 10).
Es besteht ein Zusammenhang zwischen dem PAX6-Mutationsstatus und dem Fortschreiten der AAK. Bei Patienten mit PTC- oder C-terminalen Verlängerungsmutationen schreitet die AAK altersabhängig fort, während andere Mutationstypen eine nicht-progressive Keratopathie verursachen können 11).
Das Gillespie-Syndrom wird durch Mutationen im ITPR1-Gen verursacht 3). ITPR1 gehört zur Familie der IP3-Rezeptoren, bildet Ca²⁺-Freisetzungskanäle und ist im endoplasmatischen Retikulum lokalisiert. Dominant-negative Mutationen beeinträchtigen die Bildung und Aufrechterhaltung des Irissphinkters und führen zu einer spezifischen Iris-Hypoplasie um die Pupille und einer fixierten Mydriasis.
In der Literaturübersicht zum Gillespie-Syndrom von Ciaccio et al. (2024) wurde anhand von 33 molekular bestätigten Fällen gezeigt, dass die motorische Entwicklung zwar verzögert ist, sich aber im Laufe der Zeit verbessert, dass eine geistige Behinderung nicht bei allen Fällen vorliegt und 17% eine normale Intelligenz aufweisen, und dass die neurologischen Zeichen nicht progredient sind 3).
Durch die Verbreitung der Exom-Sequenzierung werden weiterhin neue PAX6-Mutationen identifiziert. In der Human PAX6 Mutation Database waren 2018 491 Mutationen registriert, seitdem wurden etwa 250 weitere neue Mutationen berichtet1). Auch Mutationen in nicht-kodierenden Regionen werden zunehmend als Ursache für Aniridie identifiziert, was zur Aufklärung von Fällen beitragen könnte, die mit herkömmlichen Tests nicht diagnostiziert werden konnten9).
Bei Kataraktoperationen in Fällen mit schwerer AAK ist die Visualisierungstechnik mit Chandelier-Gegenbeleuchtung hilfreich4). Dieses Verfahren ermöglicht eine sichere Phakoemulsifikation auch bei Patienten mit hochgradiger AAK (Grad 3–4) und führt zu einer postoperativen Sehverbesserung.
Es zeigt sich zunehmend, dass die Progressionsmuster der AAK je nach Art der PAX6-Mutation variieren. Durch sinkende Kosten der Gentests wird die Vorhersage des klinischen Verlaufs auf Basis des Mutationstyps und eine frühzeitige Intervention zunehmend zu einer realistischen Option.
In Fällen mit gleichzeitigem Auftreten von Aniridie und Trisomie 21 wurde trotz des Vorliegens beider Erkrankungen ein relativ milder Verlauf berichtet2). Das Verständnis der Auswirkungen mehrerer genetischer Störungen auf den Phänotyp bei einem Patienten könnte wichtige Erkenntnisse für die personalisierte Medizin liefern.
Die Anwendung von Read-through-Wirkstoffen (Ataluren) bei PTC-Mutationen wird auf präklinischer Ebene für die Aniridie untersucht8). Zur PAX6-Gentherapie laufen präklinische Studien zur Genersatztherapie mit AAV-PAX6-Vektoren im Sey-Mausmodell. Künftige klinische Studien werden erwartet.
Klinische Studien zur Transplantation von iPS-Zell-abgeleiteten Hornhautepithelzellschichten werden im In- und Ausland durchgeführt und gelten als vielversprechende neue Therapie für AAK8). Für künstliche Iris (z. B. CustomFlex Artificial Iris) liegen zunehmend Erfahrungen aus dem Ausland vor. Kontaktlinsen mit künstlicher Iris sind als Hilfsmittel erstattungsfähig.
Die Erfassung der tatsächlichen Situation durch die Sammlung großer Registerdaten in Japan und die Verbesserung der Evidenzqualität sind wichtige zukünftige Aufgaben8). Eine auf individuellen Genmutationen basierende Vorhersage des AAK-Verlaufs und die Optimierung frühzeitiger Interventionen werden erwartet.