Aniridie isolée
Fréquence : environ 2/3 de l’ensemble.
Mode de transmission : autosomique dominant (AD).
Caractéristiques : due à une mutation du gène PAX6. Aucun symptôme systémique associé. Pénétrance complète mais expressivité variable.

L’aniridie est une maladie congénitale rare caractérisée par une hypoplasie ou une absence variable de l’iris. Le terme « aniridie » est un abus de langage, car des fragments de tissu irien sont presque toujours visibles à la gonioscopie ou à l’échographie biomicroscopique (UBM).
La prévalence est d’environ 1/40 000 à 1/100 000, sans différence significative de race ou de sexe rapportée 1). Dans la CIM-10, elle est classée sous Q13.1.
Il s’agit d’une maladie pan-oculaire affectant non seulement l’iris, mais aussi la cornée, le cristallin, l’angle iridocornéen, la fovéa et le nerf optique 1), entraînant diverses complications oculaires menaçant la vision. Le pronostic visuel est généralement mauvais, avec une acuité visuelle corrigée souvent limitée à environ 0,1. Le réflexe pupillaire est absent, mais l’accommodation est préservée ; 60 à 90 % des cas sont bilatéraux.
Les trois phénotypes suivants sont reconnus.
Aniridie isolée
Fréquence : environ 2/3 de l’ensemble.
Mode de transmission : autosomique dominant (AD).
Caractéristiques : due à une mutation du gène PAX6. Aucun symptôme systémique associé. Pénétrance complète mais expressivité variable.
Syndrome WAGR
Fréquence : une partie des cas sporadiques.
Mode de transmission : délétion contiguë de PAX6 et WT1.
Caractéristiques : associé à une tumeur de Wilms, des anomalies urogénitales et un retard mental. Le risque tumoral peut atteindre 50 %.
Syndrome de Gillespie
Fréquence : environ 2 % de l’ensemble.
Mode de transmission : mutation du gène ITPR1.
Caractéristiques : associé à une ataxie cérébelleuse et une déficience intellectuelle. Une anomalie irienne spécifique avec mydriase fixe est caractéristique3).
L’aniridie sporadique représente environ 1/3 de l’ensemble et est causée par une délétion de novo en 11p13 incluant PAX6. Si la délétion s’étend au gène WT1 adjacent, elle est responsable du syndrome WAGR1). 25 à 30 % des aniridies sporadiques développent une tumeur de Wilms, avec un risque relatif rapporté de 67.
PAX6 est un gène maître du développement oculaire, impliqué dans le développement de l’œil, du tube neural, du bulbe olfactif, des îlots de Langerhans du pancréas et de l’épithélium olfactif. La perte de fonction d’un allèle (haploinsuffisance) provoque la maladie ; une anomalie des deux allèles est létale in utero. En 2017, elle a été désignée maladie rare au titre de la loi sur les maladies rares, et les patients de sévérité de grade III ou plus (voir la section Diagnostic et examens pour les détails) sont éligibles à une aide financière médicale7).
Les cas sporadiques (nouvelle mutation) représentent environ un tiers de tous les cas et peuvent survenir sans antécédents familiaux. En cas de forme sporadique, un syndrome WAGR est possible, d’où l’importance d’un test génétique et d’un dépistage du néphroblastome par échographie abdominale.
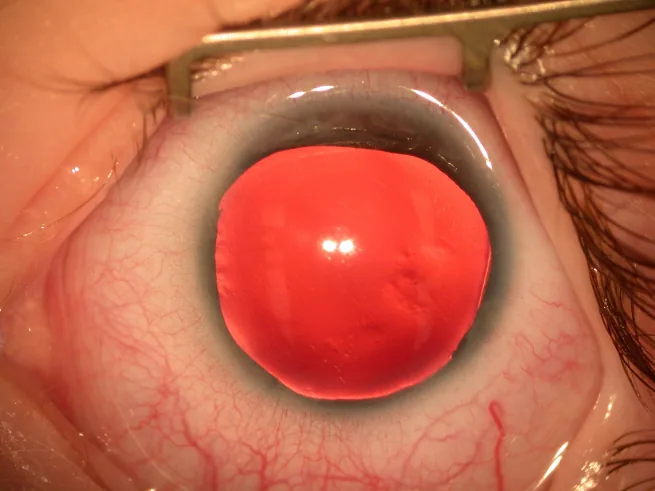
L’aniridie est souvent découverte à la naissance en raison d’une anomalie de l’iris ou de la pupille, ou dans la petite enfance à cause d’un nystagmus.
Le phénotype varie entre les familles et au sein d’une même famille, mais les différences entre les deux yeux sont généralement faibles.
En raison principalement d’une hypoplasie fovéolaire, l’acuité visuelle corrigée est souvent de l’ordre de 0,1 à 0,2. Le pronostic visuel est particulièrement mauvais en cas d’hypoplasie maculaire associée. La correction réfractive et les soins de basse vision dès la petite enfance sont importants pour le développement visuel.
PAX6 étant exprimé non seulement dans les tissus oculaires mais aussi dans le système nerveux central, les îlots de Langerhans du pancréas et l’épithélium olfactif, les complications extraoculaires suivantes peuvent être observées 8).
Les facteurs importants déterminant la fonction visuelle sont le glaucome, l’hypoplasie maculaire, le nystagmus, la kératopathie, la cataracte et l’anomalie de l’iris. La perte du champ visuel et de l’acuité visuelle due au glaucome étant irréversible, la gestion de la pression intraoculaire est primordiale lors du suivi.8)
La majorité des aniridies congénitales sont causées par des mutations hétérozygotes du gène PAX6 situé sur le bras court du chromosome 11 (11p13). L’haploinsuffisance de PAX6 est le principal mécanisme pathogène.1)
Le gène PAX6 est un gène maître du développement oculaire et joue un rôle crucial dans le développement de l’œil, du tube neural, du bulbe olfactif et du pancréas. Deux copies de PAX6 sont nécessaires au développement oculaire normal, et la perte de fonction d’une seule copie suffit à provoquer l’aniridie.1)
Dans une étude de cohorte de patients chinois, des mutations causales du gène PAX6 ont été identifiées dans 96,9 % des cas.1) Dans l’aniridie typique, des mutations induisant une dégradation de l’ARNm dépendante des mutations non-sens (NMD) ou des délétions de grande taille sont détectées dans 96 % des cas.1)
Sur le plan pathologique, on observe une absence de muscle lisse en dehors de la racine de l’iris et un développement anormal de l’angle iridocornéen. Une dysfonction des cellules souches épithéliales cornéennes entraîne des anomalies de l’épithélium et de la membrane de Bowman, avec formation d’un pannus richement vascularisé.
La répartition des mutations de PAX6 à l’origine du phénotype d’aniridie est présentée ci-dessous.
| Type de mutation | Fréquence |
|---|---|
| Mutation non-sens | Environ 39 % |
| Mutation de décalage du cadre de lecture | Environ 25 % |
| Mutation d’épissage | Environ 13 % |
| Mutation faux-sens | Environ 12 % |
Les mutations de type run-on (mutations d’extension C-terminale) représentent environ 5 % des cas, où le codon stop est converti en codon de traduction, produisant une protéine PAX6 anormalement longue 6). Les mutations d’extension C-terminale sont souvent associées à une hypoplasie irienne sévère et à une déficience visuelle importante 1)6).
Les mutations génétiques sont principalement de type PTC, mais des mutations faux-sens ont également été rapportées 7). Concernant l’utilité des tests génétiques, le séquençage Sanger ou NGS détecte des mutations chez près de 85 % des cas d’aniridie isolée. De plus, la MLPA ou l’CMA détecte des délétions dans le gène PAX6 ou dans les régions cis-régulatrices chez près de 15 % des cas 8).
Wang (2023) a identifié une nouvelle mutation de décalage du cadre de lecture c.640_646del (p.R214Pfs*28) et a rapporté un cas présentant une absence complète d’iris, une hypoplasie fovéale, une ectopie du cristallin et un décollement de la rétine 1).
Ratna et al. (2022) ont identifié une mutation run-on c.1268A>T (p.*423L) dans une famille indienne. Les personnes atteintes présentaient une aniridie complète, un nystagmus, une hypoplasie fovéale, une AAK, une subluxation supérieure du cristallin, une myopie forte et une atrophie optique, illustrant le phénotype sévère associé aux mutations d’extension C-terminale 6).
Dans l’aniridie sporadique, les grandes délétions incluant le gène WT1 en plus de PAX6 sont responsables du syndrome WAGR. Le risque de tumeur de Wilms en cas de délétion de WT1 peut atteindre 50 % 1). En cas de suspicion de syndrome WAGR, des tests génétiques doivent confirmer les délétions de PAX6 et WT1, permettant d’évaluer le risque de tumeur de Wilms et de suivre les retards de développement 8). L’évaluation de la région WT1 par tests génétiques est essentielle, car 30 % des cas sporadiques développent une tumeur de Wilms avant l’âge de 5 ans. La proximité des gènes WT1 et PAX6 explique que la délétion du bras court du chromosome 11 (délétion 11p13) entraîne à la fois une aniridie et une tumeur de Wilms.
Le syndrome de Gillespie est causé par des mutations hétérozygotes dominantes négatives ou bialléliques du gène ITPR1 3). À ce jour, 37 cas ont été confirmés par diagnostic moléculaire, et le résidu Gly2554 est connu comme un point chaud 3).
Selon les critères diagnostiques de l’aniridie (2020), le diagnostic est confirmé sur la base des éléments suivants 7).
A. Symptômes
B. Résultats d’examen
C. Diagnostics différentiels
E. Tests génétiques : mutation pathogène du gène PAX6 ou délétion de la région 11p13
Catégories diagnostiques7) :
La classification de la sévérité pour la reconnaissance comme maladie rare est définie en quatre stades7).
| Sévérité | Définition |
|---|---|
| Stade I | Atteinte unilatérale, œil controlatéral sain |
| Degré II | Atteinte bilatérale, meilleure acuité visuelle corrigée ≥ 0,3 |
| Degré III | Atteinte bilatérale, meilleure acuité visuelle corrigée ≥ 0,1 et < 0,3 |
| Degré IV | Atteinte bilatérale, meilleure acuité visuelle corrigée < 0,1 |
Même pour les degrés I à III, en cas de rétrécissement du champ visuel dû à un glaucome (champ visuel central résiduel ≤ 20° avec la cible Goldmann I/4), le degré de sévérité est augmenté d’un niveau. Un degré de sévérité ≥ III ouvre droit à une aide financière médicale7).
Le diagnostic clinique est facile si l’on observe une absence ou une hypoplasie de l’iris à la lampe à fente. L’évaluation du tissu irien résiduel se fait par gonioscopie ou microscopie ultrasonique biomicroscopique. On vérifie également la présence d’anomalies de développement de l’angle de la chambre antérieure.
Les complications oculaires suivantes sont évaluées systématiquement :
L’objectif le plus important de l’évaluation génétique de l’aniridie est de confirmer si la délétion de PAX6 s’étend au gène WT11). L’analyse de l’exome entier ou la technique MLPA permettent d’évaluer les mutations et délétions dans les régions PAX6 et WT11)2).
Dans l’aniridie sporadique, l’évaluation du risque de tumeur de Wilms par délétion du gène WT1 est directement liée au pronostic vital1). Même dans les formes familiales, en raison de la diversité phénotypique, un diagnostic génétique et un conseil génétique sont recommandés.
Il n’existe pas de traitement curatif de l’aniridie. La prise en charge repose sur les soins de basse vision pour maximiser l’utilisation de la vision résiduelle et le traitement individuel de chaque complication8).
La greffe de cornée pour l’opacité du stroma cornéen doit être envisagée avec prudence8).
La greffe de cornée peut améliorer la vision à court terme, mais l’amélioration est limitée en raison de comorbidités telles que l’hypoplasie maculaire. À long terme, la progression du glaucome et la dysfonction du greffon entraînent un mauvais pronostic visuel.
En cas d’insuffisance de cellules souches épithéliales cornéennes, envisager un traitement chirurgical8).
La chirurgie de la cataracte doit être envisagée en fonction du degré d’opacité et de photophobie8).
La cataracte survient chez 50 à 85 % des patients avant l’âge de 20 ans. La chirurgie est planifiée en fonction de l’intensité de l’opacité et de la photophobie. Une amélioration visuelle a été rapportée dans 66 à 100 % des cas opérés, mais les points suivants nécessitent une attention particulière.
En raison de la fragilité des zonules de Zinn, l’implantation d’un cristallin artificiel nécessite une indication prudente.
Hu et al. (2024) ont réalisé une phacoémulsification assistée par rétroéclairage chandelier chez deux patients atteints d’aniridie congénitale avec AAK sévère. Bien que la visualisation peropératoire standard fût difficile en raison de l’opacité cornéenne, l’éclairage postérieur a permis une visualisation claire du cristallin et de la capsule antérieure, avec une amélioration de l’acuité visuelle corrigée à 20/200 et 20/1000 respectivement à 3 semaines postopératoires 4).
Le glaucome étant directement lié au pronostic visuel, il doit être traité activement 8).
Après l’apparition du glaucome, la prise en charge suit un algorithme en 5 étapes.
Traitement médicamenteux : Les bêta-bloquants, les sympathicomimétiques et les prostaglandines (PG) sont efficaces. La brimonidine (agoniste alpha-adrénergique) est contre-indiquée chez les enfants de moins de 2 ans en raison du risque de dépression du système nerveux central. En cas de risque d’atteinte épithéliale cornéenne, utiliser des préparations sans conservateur.
Chirurgie reconstructrice de la voie d’écoulement (goniotomie, trabéculotomie) : Recommandée comme première intervention 16). Des goniotomies prophylactiques ont également été rapportées. Cependant, elle peut être inefficace si l’iris résiduel recouvre le trabéculum.
Chirurgie filtrante (trabéculectomie) : Seulement quelques rapports à court et moyen terme. Les résultats sont souvent médiocres chez l’enfant, avec une hypotonie postopératoire dans environ 25 % des cas 13). Des cas de glaucome malin postopératoire ont également été rapportés.
Chirurgie d’implant de glaucome (tube shunt) : Les dispositifs de type Baerveldt ou Ahmed peuvent être utilisés. En cas d’œil phaque, l’insertion du tube doit être tangentielle plutôt que dirigée vers le centre cornéen. Un bon contrôle de la pression intraoculaire est attendu.
Cyclocoagulation : Dernier recours. La cryocoagulation du corps ciliaire a souvent conduit à une hypotonie. En raison de l’hypoplasie ciliaire, le risque d’hypotonie est plus élevé que dans un œil sain.
En raison de l’anomalie de développement de l’angle, une approche différente de celle du glaucome à angle ouvert habituel est nécessaire. En première intention, on choisit la chirurgie reconstructrice de la voie d’écoulement, puis la chirurgie de shunt tubulaire constitue une bonne option. La brimonidine est contre-indiquée chez les enfants de moins de 2 ans, et l’utilisation d’antimétabolites peut aggraver l’AAK, nécessitant une décision prudente 8).
Les soins pour basse vision doivent être introduits précocement8).
La correction de la réfraction est fondamentale, le taux de complication de myopie étant supérieur à 64 %.
Le traitement de la photophobie est important pour préserver le développement de la fonction visuelle et la qualité de vie8).
La plupart des patients peuvent fréquenter une classe ordinaire, mais des aides telles que des manuels agrandis, des tablettes et des pupitres inclinés sont nécessaires. L’intégration dans une classe pour malvoyants ou le recours aux consultations parentales et éducatives dans les écoles pour aveugles ou les écoles spécialisées pour déficients visuels sont également des options.
Depuis avril 2017, cette maladie est reconnue comme maladie rare désignée. Ainsi, même sans carte de handicap, les patients de sévérité de grade III ou plus peuvent bénéficier d’une aide financière pour les frais médicaux et de subventions pour les appareils d’assistance 7). Les appareils concernés comprennent les lunettes correctrices, les lunettes filtrantes, les lentilles de contact (y compris avec iris artificiel), les loupes pour basse vision, les cannes de sécurité pour déficients visuels et les prothèses oculaires.
PAX6 s’étend sur 22 kb d’ADN génomique comprenant 14 exons et code pour 422 acides aminés 1). Il possède deux domaines de liaison à l’ADN (domaine apparié et homéodomaine apparié), et le domaine PST (riche en proline, sérine et thréonine) en C-terminal agit comme activateur transcriptionnel.
PAX6 régule la prolifération, la différenciation, la migration et l’adhésion cellulaires, et ses cibles incluent PAX6 lui-même ainsi que les gènes codant pour les cristallines du cristallin et les kératines cornéennes. L’expression se poursuit dans la rétine, le cristallin et la cornée adultes. PAX6 est l’un des gènes maîtres contrôlant la différenciation des organes embryonnaires.
La plupart des mutations de PAX6 provoquent une haploinsuffisance via la dégradation des ARNm non-sens (NMD) 1). Les mutations introduisant un codon stop prématuré (PTC) (mutations non-sens, décalages du cadre de lecture, et la plupart des mutations d’épissage) entraînent un phénotype typique d’aniridie.
En revanche, si le PTC est situé dans le dernier exon ou dans les 50 pb terminales de l’avant-dernier exon, il peut échapper au NMD, et une protéine tronquée peut être traduite, conduisant à un phénotype sévère 1).
Un cas rare de mutation non-sens de PAX6 c.282C>A (p.Cys94*) associée à une trisomie 21 chez un même patient a été rapporté. La mutation PAX6 est survenue de novo, entraînant une aniridie bilatérale complète, un glaucome congénital, un AAK et une hypoplasie fovéolaire 2).
Bien qu’aucune corrélation génotype-phénotype claire ne soit établie, certaines tendances sont connues 1).
Dans la série de gonioscopies de Grant et Walton, il a été montré que le stroma irien s’étend initialement vers l’avant sur le trabéculum, formant des adhérences, puis devient progressivement en feuillet et conduit finalement à une occlusion de l’angle14). Ce mécanisme est le facteur principal du développement du glaucome. Sur le plan pathologique, un défaut du muscle lisse avec préservation de la racine de l’iris et un développement insuffisant de l’angle sont à la base.
L’AAK est principalement causée par une déficience en cellules souches limbiques (LSCD), mais une différenciation anormale de l’épithélium cornéen, des anomalies d’adhésion, une infiltration de cellules conjonctivales et une production insuffisante de larmes sont également impliquées. Un déficit en métalloprotéinase matricielle 9 (MMP-9), régulée par PAX6, entraîne une accumulation de fibrine et une infiltration de cellules inflammatoires, et la perte de transparence résulte d’un désordre de la séquence de collagène du stroma.
L’AAK est classée en 5 stades. Le stade I ne présente qu’une anomalie de l’épithélium périphérique, le stade II un changement épithélial centripète (n’atteignant pas le centre), le stade III un changement épithélial cornéen central et une néovascularisation superficielle périphérique, le stade IV une néovascularisation superficielle de toute la cornée, et le stade V une anomalie épithéliale de toute la cornée et une cicatrice stromale profonde10).
Il existe une relation entre le statut de mutation de PAX6 et la progression de l’AAK. Chez les patients présentant des mutations PTC ou d’extension C-terminale, l’AAK progresse de manière dépendante de l’âge, tandis que d’autres types de mutations peuvent entraîner une kératopathie non progressive11).
Le syndrome de Gillespie est causé par des mutations du gène ITPR13). ITPR1 fait partie de la famille des récepteurs IP3, forme un canal de libération de Ca²⁺ et est localisé dans le réticulum endoplasmique. Les mutations dominantes négatives affectent la formation et le maintien du sphincter irien, entraînant une hypoplasie irienne spécifique autour de la pupille et une mydriase fixe.
Dans une revue de la littérature sur le syndrome de Gillespie par Ciaccio et al. (2024), l’analyse de 33 cas confirmés moléculairement a montré que le développement moteur est retardé mais s’améliore avec le temps, que la déficience intellectuelle n’est pas présente dans tous les cas (17% ont une intelligence normale) et que les signes neurologiques sont non progressifs3).
Grâce à la généralisation du séquençage de l’exome entier, de nouvelles mutations de PAX6 sont continuellement identifiées. En 2018, la Human PAX6 Mutation Database comptait 491 mutations, et environ 250 nouvelles mutations ont été signalées depuis 1). Des mutations dans les régions non codantes sont également identifiées comme causes d’aniridie, ce qui devrait permettre d’élucider des cas qui n’étaient pas diagnostiqués par les tests conventionnels 9).
Pour la chirurgie de la cataracte chez les patients présentant une AAK sévère, la technique de visualisation assistée par éclairage chandelier inversé est utile 4). Cette technique permet une phacoémulsification sûre même chez les patients avec une AAK de grade 3 à 4, et une amélioration de l’acuité visuelle postopératoire a été obtenue.
Il devient clair que le type de mutation de PAX6 influence le schéma de progression de l’AAK. Avec la baisse du coût des tests génétiques, la prédiction de l’évolution clinique basée sur le type de mutation et l’intervention précoce deviennent des options réalistes.
Dans les cas d’association d’aniridie et de trisomie 21, des exemples d’évolution relativement bénigne malgré la coexistence des deux maladies ont été rapportés 2). Comprendre l’impact sur le phénotype lorsque plusieurs troubles génétiques coexistent chez un même patient pourrait apporter des connaissances importantes pour la médecine personnalisée future.
L’application de l’ataluren, un médicament de lecture forcée pour les mutations de type PTC, à l’aniridie est étudiée au niveau de la recherche fondamentale 8). Concernant la thérapie génique de PAX6, des recherches fondamentales sur le remplacement génique par un vecteur AAV-PAX6 dans un modèle murin de mutation Sey sont en cours. Le développement vers des essais cliniques futurs est attendu.
Des essais de transplantation de feuillets de cellules épithéliales cornéennes dérivées de cellules iPS sont menés au Japon et à l’étranger, et cette technique est considérée comme une nouvelle option thérapeutique pour l’AAK 8). Pour les iris artificiels (comme le CustomFlex Artificial Iris), l’expérience clinique à l’étranger s’accumule. Les lentilles de contact avec iris artificiel sont prises en charge par l’assurance maladie en tant qu’appareils de compensation.
L’accumulation de données de registre à grande échelle au Japon pour comprendre la situation réelle et améliorer la qualité des preuves est un enjeu important pour l’avenir 8). On espère optimiser la prédiction de la progression de l’AAK et l’intervention précoce basées sur les mutations génétiques individuelles.