Isolated Aniridia
Frequency: Approximately 2/3 of all cases.
Inheritance: Autosomal dominant (AD).
Features: Caused by PAX6 gene mutation. No systemic symptoms. Penetrance is complete but expressivity is variable.

Aniridia is a rare congenital disorder characterized by varying degrees of iris hypoplasia or absence. The term “aniridia” is a misnomer, as remnants of iris tissue are almost always detectable by gonioscopy or ultrasound biomicroscopy (UBM).
The prevalence is approximately 1 in 40,000 to 1 in 100,000, with no significant racial or sex differences reported 1). It is classified under ICD-10 code Q13.1.
Aniridia is a panocular disease affecting not only the iris but also the cornea, lens, angle, fovea, and optic nerve 1), presenting various vision-threatening ocular complications. Visual prognosis is generally poor, with corrected visual acuity often around 0.1. Pupillary reflexes are absent, but accommodative responses are preserved, and 60–90% of cases are bilateral.
The following three phenotypes are recognized.
Isolated Aniridia
Frequency: Approximately 2/3 of all cases.
Inheritance: Autosomal dominant (AD).
Features: Caused by PAX6 gene mutation. No systemic symptoms. Penetrance is complete but expressivity is variable.
WAGR Syndrome
Frequency: A subset of sporadic cases.
Inheritance: Contiguous deletion of PAX6 and WT1.
Features: Associated with Wilms tumor, genitourinary abnormalities, and intellectual disability. Tumor risk is up to 50%.
Gillespie Syndrome
Frequency: Approximately 2% of all cases.
Inheritance: ITPR1 gene mutation.
Features: Accompanied by cerebellar ataxia and intellectual disability. Characteristic iris abnormality with fixed dilated pupils is distinctive3).
Sporadic aniridia accounts for about 1/3 of all cases and is caused by a de novo deletion of 11p13 including PAX6. If the deletion extends to the adjacent WT1 gene, it causes WAGR syndrome1). 25–30% of sporadic aniridia patients develop Wilms tumor, with a reported relative risk of 67.
PAX6 is a master control gene for eye formation and is involved in the development of the eye, neural tube, olfactory bulb, pancreatic islets of Langerhans, and olfactory epithelium. It manifests with loss of function of one allele (haploinsufficiency), and biallelic abnormalities are embryonic lethal. In 2017, it was designated as a specified intractable disease under the Intractable Disease Act, and patients with severity grade III or higher (see Diagnosis and Testing section for details) are eligible for medical expense subsidies7).
Sporadic (new mutation) cases account for about one-third of all cases and can occur without a family history. Since sporadic cases may indicate WAGR syndrome, genetic testing and abdominal ultrasound screening for Wilms tumor are important.
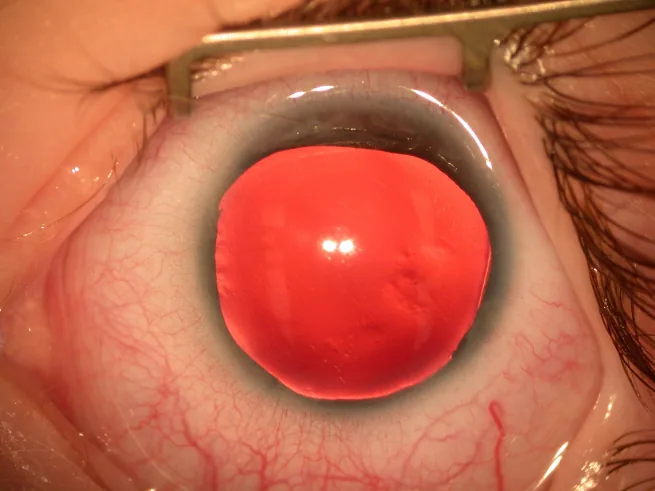
Most cases of aniridia are detected at birth due to iris and pupil abnormalities, or in infancy due to nystagmus.
The phenotype varies between and within families, but differences between the right and left eyes are usually small.
Due primarily to foveal hypoplasia, corrected visual acuity is often around 0.1 to 0.2 (20/200 to 20/100). Visual prognosis is particularly poor when macular hypoplasia is present. Refractive correction and low vision care from infancy are important for visual development.
PAX6 is expressed not only in ocular tissues but also in the central nervous system, pancreatic islets of Langerhans, and olfactory epithelium, so the following extraocular complications may occur 8).
Key factors determining visual function are glaucoma, macular hypoplasia, nystagmus, keratopathy, cataract, and iris hypoplasia. Since visual field and acuity loss due to glaucoma are irreversible, intraocular pressure management is the most important aspect of follow-up8).
Most cases of congenital aniridia are caused by heterozygous mutations in the PAX6 gene located on the short arm of chromosome 11 (11p13). Haploinsufficiency is the main mechanism of PAX6-related disease1).
The PAX6 gene is a master control gene for eye formation and plays important roles in the development of the eye, neural tube, olfactory bulb, and pancreas. Two copies of PAX6 are required for normal eye development, and loss of function of just one copy leads to aniridia1).
In a cohort study of Chinese patients, causative mutations in the PAX6 gene were identified in 96.9% of cases1). In typical aniridia, mutations that induce nonsense-mediated mRNA decay (NMD) or large deletions are detected in 96% of cases1).
Pathologically, smooth muscle is absent except at the iris root, and there is developmental abnormality of the angle. Functional abnormalities of corneal epithelial stem cells lead to abnormalities in the epithelium and Bowman’s layer, with formation of a vascularized pannus.
The breakdown of PAX6 mutations causing the aniridia phenotype is shown below.
| Mutation type | Frequency |
|---|---|
| Nonsense mutation | Approximately 39% |
| Frameshift mutation | Approximately 25% |
| Splice site mutation | Approximately 13% |
| Missense mutation | Approximately 12% |
Run-on mutations (C-terminal extension mutations) account for about 5% and produce an abnormally elongated PAX6 protein due to conversion of a stop codon into a coding codon 6). C-terminal extension mutations are often associated with severe iris hypoplasia and profound visual impairment 1)6).
Gene mutations are often of the PTC type, and missense mutations have also been reported 7). Regarding the utility of genetic testing, mutations are detected in nearly 85% of isolated aniridia cases by Sanger sequencing or NGS. Additionally, deletions within the PAX6 gene or its cis-regulatory region are detected in nearly 15% of cases by MLPA or CMA 8).
Wang (2023) newly identified a frameshift mutation c.640_646del (p.R214Pfs*28) and reported a case presenting with complete iris defect, foveal hypoplasia, lens subluxation, and retinal detachment 1).
Ratna et al. (2022) identified a run-on mutation c.1268A>T (p.*423L) in an Indian family. Affected individuals presented with complete aniridia, nystagmus, foveal hypoplasia, AAK, superior lens subluxation, high myopia, and optic atrophy, demonstrating a severe phenotype due to C-terminal extension mutation 6).
In sporadic aniridia, large deletions involving the WT1 gene in addition to PAX6 cause WAGR syndrome. The risk of Wilms tumor in cases with WT1 deletion is up to 50% 1). If WAGR syndrome is suspected, genetic testing can confirm PAX6 and WT1 deletions, enabling Wilms tumor risk assessment and monitoring for developmental delay 8). Evaluation of the WT1 region by genetic testing is essential, and 30% of sporadic cases develop Wilms tumor by age 5. Because the WT1 gene is located near PAX6, deletion of both on chromosome 11 short arm (11p13 deletion) results in aniridia complicated by Wilms tumor.
Gillespie syndrome is caused by heterozygous dominant-negative mutations or biallelic mutations in the ITPR1 gene 3). To date, 37 cases with molecular diagnosis have been reported, and the Gly2554 residue is known as a hotspot 3).
Based on the diagnostic criteria for aniridia (2020), the diagnosis is confirmed according to the following criteria7).
A. Symptoms
B. Examination Findings
C. Differential Diagnoses
E. Genetic testing: Pathogenic mutation in the PAX6 gene or deletion of the 11p13 region
Diagnostic categories7):
The severity classification for intractable disease designation is defined in the following four stages7).
| Severity | Definition |
|---|---|
| Grade I | Unilateral involvement, contralateral eye normal |
| Grade II | Both eyes affected, corrected visual acuity in the better eye 0.3 or better |
| Grade III | Both eyes affected, corrected visual acuity in the better eye 0.1 or better but less than 0.3 |
| Grade IV | Both eyes affected, corrected visual acuity in the better eye less than 0.1 |
Even for grades I to III, if accompanied by visual field narrowing due to glaucoma or other conditions (central residual visual field within 20 degrees on Goldmann I/4 target), the severity shifts up by one grade. Severity grade III or higher is eligible for medical expense subsidies 7).
Clinical diagnosis is easy if iris defects or hypoplasia are confirmed by slit-lamp microscopy. Gonioscopy or ultrasound biomicroscopy is used to evaluate residual iris tissue. The presence or absence of developmental abnormalities of the anterior chamber angle is also checked.
The following ocular complications are systematically evaluated:
The most important goal in the genetic evaluation of aniridia is to confirm whether the PAX6 deletion extends to the WT1 gene 1). Whole-exome sequencing and MLPA are used to evaluate mutations and deletions in the PAX6 and WT1 regions 1)2).
In sporadic aniridia, assessment of Wilms tumor risk due to WT1 gene deletion is directly linked to prognosis 1). Even in familial cases, due to phenotypic variability, genetic testing for definitive diagnosis and genetic counseling is recommended.
There is no curative treatment for aniridia. Management focuses on low-vision care to maximize residual vision and individual treatment for each complication 8).
Corneal transplantation for stromal opacification should be considered cautiously 8).
Corneal transplantation may provide short-term visual improvement, but improvement is limited due to concurrent conditions such as macular hypoplasia. Long-term visual prognosis is poor due to progression of glaucoma and graft failure.
Surgical treatment should be considered for corneal epithelial stem cell deficiency8).
Cataract surgery should be considered based on the degree of opacity and photophobia8).
Cataracts develop in 50–85% of patients by age 20. Surgery is planned according to the severity of opacity and photophobia. Visual improvement has been reported in 66–100% of surgical cases, but the following points require attention.
Because the zonules of Zinn are fragile, intraocular lens insertion requires careful consideration.
Hu et al. (2024) performed phacoemulsification assisted by chandelier retroillumination in two cases of congenital aniridia with severe AAK. Although conventional intraoperative visualization was difficult due to corneal opacity, posterior illumination enabled clear visualization of the lens and anterior capsule, and corrected visual acuity improved to 20/200 and 20/1000 at 3 weeks postoperatively 4).
Glaucoma is directly linked to visual prognosis, so aggressive treatment is necessary 8).
After the onset of glaucoma, management is based on the following 5-step algorithm.
Medical therapy: Beta-blockers, sympathomimetics, and prostaglandin (PG) analogs are effective. Brimonidine (alpha-adrenergic agonist) is contraindicated in children under 2 years due to risk of central nervous system depression. If corneal epithelial damage is a concern, use preservative-free formulations.
Outflow reconstruction surgery (goniotomy/trabeculotomy): Recommended as the initial surgery 16). Prophylactic goniotomy has also been reported. However, it may be ineffective in cases where residual iris covers the trabecular meshwork.
Filtering surgery (trabeculectomy): Only a few short- to medium-term reports exist. Outcomes tend to be poor in pediatric eyes, with postoperative ocular hypotony occurring in about 25% of cases 13). Postoperative malignant glaucoma has also been reported.
Glaucoma implant surgery (tube shunt surgery): Baerveldt and Ahmed devices are available. For phakic eyes, tube insertion should be directed tangentially rather than toward the center of the cornea. Good intraocular pressure control can be expected.
Cyclodestructive procedures: Last resort. Cyclocryotherapy has been reported to result in ocular hypotony in many cases. Due to ciliary body hypoplasia, the risk of hypotony is higher than in normal eyes.
Because the underlying cause is an abnormality in the development of the angle, a different approach from that for typical open-angle glaucoma is necessary. The first choice is outflow reconstruction surgery, followed by tube shunt surgery as a good option. Brimonidine is contraindicated in children under 2 years of age, and the use of antimetabolites may worsen AAK, so careful judgment is required 8).
Low vision care should be introduced early 8).
Refractive correction is fundamental, and the rate of myopia complication is reported to be 64% or higher.
Treatment of photophobia is important for maintaining visual function development and quality of life 8).
Most patients can attend regular classes, but support such as enlarged textbooks, tablet devices, and book stands is necessary. Options include attending classes for visually impaired students or using childcare and educational consultations at schools for the blind or special support schools for visual impairments.
Since April 2017, this condition has been designated as a specified intractable disease. Therefore, even without a physical disability certificate, patients with severity level III or higher are eligible for medical expense subsidies and assistive device provision 7). Eligible assistive devices include corrective glasses, tinted glasses, contact lenses (including those with artificial iris), low-vision glasses, white canes for the visually impaired, and artificial eyes.
PAX6 spans 22 kb of genomic DNA containing 14 exons and encodes 422 amino acids 1). It has two DNA-binding domains (paired domain and paired-type homeodomain), and the C-terminal PST (proline-serine-threonine-rich) domain functions as a transcriptional activator.
PAX6 regulates cell proliferation, differentiation, migration, and adhesion. Its targets include PAX6 itself as well as genes encoding lens crystallins and corneal keratins. Expression continues in the adult retina, lens, and cornea. PAX6 is one of the master control genes governing organ differentiation during the embryonic period.
Most PAX6 mutations cause haploinsufficiency through nonsense-mediated mRNA decay (NMD) 1). Mutations introducing a premature termination codon (PTC) (nonsense mutations, frameshift mutations, and most splice mutations) result in the typical aniridia phenotype.
Conversely, if the PTC is located in the final exon or within the last 50 bp of the penultimate exon, it may escape NMD, leading to translation of a truncated protein and potentially a severe phenotype 1).
A rare case has been reported where a PAX6 nonsense mutation c.282C>A (p.Cys94*) and trisomy 21 coexisted in the same patient. The PAX6 mutation occurred de novo, resulting in complete bilateral aniridia, congenital glaucoma, AAK, and foveal hypoplasia 2).
Although a clear genotype–phenotype correlation has not been established, several trends are known 1).
In the gonioscopic series by Grant and Walton, the iris stroma initially extends anteriorly over the trabecular meshwork, forming adhesion-like attachments that gradually become sheet-like and eventually lead to angle closure14). This mechanism is the primary cause of glaucoma development. Pathologically, the underlying basis is a deficiency of smooth muscle sparing the iris root and developmental abnormalities of the angle.
AAK is primarily caused by limbal stem cell deficiency (LSCD), but abnormal differentiation and adhesion of corneal epithelium, conjunctival cell invasion, and insufficient tear production also contribute. Deficiency of matrix metalloproteinase 9 (MMP-9), regulated by PAX6, leads to fibrin accumulation and inflammatory cell infiltration, resulting in loss of transparency due to disorganized collagen arrangement in the stroma.
AAK is classified into five stages. Stage I shows abnormalities only in the peripheral epithelium, Stage II shows centripetal epithelial changes (not reaching the center), Stage III shows epithelial changes in the central cornea and superficial neovascularization in the periphery, Stage IV shows superficial neovascularization of the entire cornea, and Stage V shows epithelial abnormalities and deep stromal scarring of the entire cornea10).
There is a relationship between PAX6 mutation status and the progression of AAK. In patients with PTC or C-terminal extension mutations, AAK progresses in an age-dependent manner, whereas other mutation types may present with non-progressive keratopathy11).
Gillespie syndrome is caused by mutations in the ITPR1 gene3). ITPR1 is a member of the IP3 receptor family, forming Ca²⁺ release channels and localizing to the endoplasmic reticulum. Dominant-negative mutations affect the formation and maintenance of the iris sphincter muscle, leading to characteristic iris hypoplasia around the pupil and fixed mydriasis.
In a literature review of Gillespie syndrome by Ciaccio et al. (2024), analysis of 33 molecularly confirmed cases revealed that motor development is delayed but improves over time, intellectual disability is not present in all cases (17% have normal intelligence), and neurological signs are non-progressive3).
With the widespread use of whole-exome sequencing, new PAX6 mutations continue to be identified. As of 2018, 491 mutations were registered in the Human PAX6 Mutation Database, and approximately 250 new mutations have been reported since then 1). Mutations in non-coding regions are also being identified as causes of aniridia, offering hope for elucidating cases that could not be diagnosed with conventional testing 9).
For cataract surgery in cases with severe AAK, visualization techniques using chandelier retroillumination are useful 4). This technique enables safe phacoemulsification even in patients with Grade 3–4 severe AAK, and postoperative visual improvement has been achieved.
It is becoming clear that the progression pattern of AAK differs depending on the type of PAX6 mutation. With decreasing costs of genetic testing, predicting clinical course based on mutation type and early intervention are becoming realistic options.
In cases of combined aniridia and trisomy 21, relatively mild courses have been reported despite the coexistence of both conditions 2). Understanding the impact on phenotype when multiple genetic disorders coexist in the same patient may provide important insights for future personalized medicine.
The application of read-through drugs (ataluren) for PTC-type mutations in aniridia is being investigated at the basic research level 8). Regarding PAX6 gene therapy, basic research on gene supplementation using AAV-PAX6 vectors in Sey mutant mouse models is ongoing. Future clinical trials are anticipated.
Clinical trials of iPS cell-derived corneal epithelial cell sheet transplantation are being conducted domestically and internationally, attracting attention as a new treatment for AAK 8). For artificial irises (e.g., CustomFlex Artificial Iris), clinical experience is accumulating overseas. Contact lenses with artificial irises are covered by insurance as prosthetic devices.
Accumulating large-scale registry data in Japan to understand the actual situation and improve the quality of evidence are important future tasks 8). Optimization of AAK progression prediction and early intervention based on individual genetic mutations is expected.