고립성 무홍채증
빈도: 전체의 약 2/3.
유전 양식: 상염색체 우성(AD).
특징: PAX6 유전자 변이에 의함. 전신 증상을 동반하지 않음. 침투율은 완전하지만 표현도는 다양함.

무홍채증(Aniridia)은 홍채의 다양한 정도의 형성 부전 또는 결손을 특징으로 하는 드문 선천성 질환입니다. ‘무홍채증’이라는 이름은 잘못된 명칭이며, 각경 검사나 초음파 생체현미경(UBM)에서는 홍채 조직의 조각이 거의 항상 확인됩니다.
유병률은 약 1/40,000~1/100,000으로 알려져 있으며, 현저한 인종 차이나 성별 차이는 보고되지 않았습니다1). ICD-10에서는 Q13.1로 분류됩니다.
홍채뿐만 아니라 각막, 수정체, 전방각, 중심와, 시신경에도 영향을 미치는 범안구 질환이며1), 시력을 위협하는 다양한 안과 합병증을 나타냅니다. 시력 예후는 대체로 불량하여 교정시력 0.1 정도에 머무르는 경우가 많습니다. 동공 반응은 소실되었지만 조절 반응은 유지되며, 60~90%가 양안성입니다.
다음 3가지 표현형이 인정된다.
고립성 무홍채증
빈도: 전체의 약 2/3.
유전 양식: 상염색체 우성(AD).
특징: PAX6 유전자 변이에 의함. 전신 증상을 동반하지 않음. 침투율은 완전하지만 표현도는 다양함.
WAGR 증후군
빈도: 산발성 예의 일부.
유전 양식: PAX6와 WT1의 인접 결실.
특징: Wilms 종양, 비뇨생식기 이상, 정신 지체를 동반함. 종양 위험은 최대 50%.
길레스피 증후군
빈도: 전체의 약 2%.
유전 양식: ITPR1 유전자 변이.
특징: 소뇌 실조, 지적 장애를 동반함. 고정 산동을 나타내는 특이적인 홍채 이상이 특징적3).
산발성 무홍채증은 전체의 약 1/3을 차지하며, PAX6를 포함한 11p13의 de novo 결실에 의해 발생한다. 인접한 WT1 유전자까지 결실이 미치는 경우 WAGR 증후군의 원인이 된다1). 산발성 무홍채증의 25~30%가 Wilms 종양을 발병하며, 상대 위험도는 67로 보고되었다.
PAX6는 안구 형성의 마스터 조절 유전자이며, 눈, 신경관, 후각 망울, 췌장 랑게르한스섬, 후각 상피의 발달에 관여한다. 한쪽 대립유전자의 기능 상실(반수체 불충분)로 발병하며, 양쪽 대립유전자에 이상이 있을 경우 태생기에 치명적이다. 2017년 난치병법이 정하는 지정 난치병이 되었으며, 중증도 III도 이상(자세한 내용은 진단·검사 항목 참조)에서 의료비 지원 대상이 된다7).
산발성(새로운 돌연변이)은 전체의 약 1/3을 차지하며, 가족력이 없어도 발병합니다. 산발성 예에서는 WAGR 증후군의 가능성이 있으므로 유전자 검사와 복부 초음파를 통한 Wilms 종양 선별검사가 중요합니다.
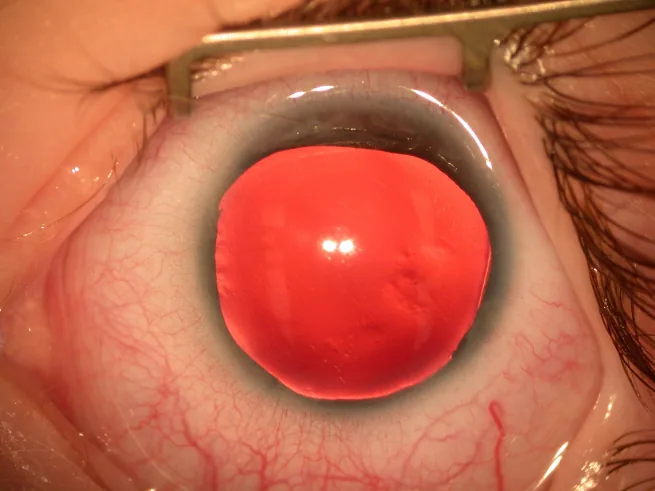
무홍채증의 대부분은 출생 시 홍채·동공 이상, 또는 유아기의 안진에 의해 발견됩니다.
표현형은 가족 간 및 가족 내에서 다르지만, 좌우안의 차이는 보통 작습니다.
중심와 형성 부전이 주된 원인이 되어, 교정 시력은 0.1~0.2 정도인 경우가 많습니다. 황반 저형성을 합병한 경우 특히 시력 예후가 불량합니다. 영유아기부터의 굴절 교정과 저시력 관리가 시각 발달에 중요합니다.
PAX6는 안조직 외에도 중추신경, 췌장 Langerhans 섬, 후각 상피에서도 발현되므로, 다음과 같은 안외 합병증이 나타날 수 있습니다8).
시력을 결정하는 중요한 인자는 녹내장, 황반 저형성, 안구진탕증, 각막증, 백내장, 홍채 형성 이상이며, 녹내장으로 인한 시야·시력 손상은 비가역적이므로 경과 관찰 시 안압 관리가 가장 중요합니다8).
대부분의 선천성 무홍채증은 11번 염색체 단완(11p13)에 위치한 PAX6 유전자의 이형접합 변이로 인해 발생합니다. PAX6는 반수체 불충분이 주요 발병 기전입니다1).
PAX6 유전자는 눈 형성의 마스터 조절 유전자로, 눈, 신경관, 후각망울, 췌장 발달에 중요한 역할을 합니다. 정상적인 눈 발달에는 2개의 PAX6 복제본이 필요하며, 하나의 기능 상실만으로도 무홍채증이 발생합니다1).
중국인 환자 코호트 연구에서는 96.9%의 증례에서 PAX6 유전자에 원인 변이가 확인되었습니다1). 전형적인 무홍채증에서는 넌센스 변이 의존 mRNA 분해(NMD)를 유도하는 변이 또는 대규모 결실이 96%에서 검출됩니다1).
병리학적으로는 홍채 뿌리를 남기고 평활근이 결손되어 있으며, 전방각 발달 부전이 관찰됩니다. 각막 상피 줄기세포의 기능 이상이 나타나 상피와 보우만막에 이상을 초래하고, 혈관이 풍부한 판누스가 형성됩니다.
무홍채증 표현형을 유발하는 PAX6 변이의 구성은 다음과 같습니다.
| 변이 유형 | 빈도 |
|---|---|
| 넌센스 변이 | 약 39% |
| 프레임시프트 변이 | 약 25% |
| 스플라이스 변이 | 약 13% |
| 미스센스 돌연변이 | 약 12% |
런온 돌연변이(C-말단 연장 돌연변이)는 약 5%를 차지하며, 종결코돈이 번역코돈으로 전환되어 비정상적으로 연장된 PAX6 단백질이 생성됩니다6). C-말단 연장 돌연변이는 중증의 홍채형성부전과 고도의 시력 장애를 동반하는 경우가 많습니다1)6).
유전자 돌연변이는 PTC형 돌연변이가 많고, 미스센스 돌연변이의 보고도 있습니다7). 유전학적 검사의 유용성에 대해, Sanger 시퀀싱 또는 NGS로 isolated aniridia의 85% 가까이에서 돌연변이가 검출됩니다. 또한 MLPA 또는 CMA로 PAX6 유전자 내 또는 cis-조절 영역의 결손이 15% 가까이 검출됩니다8).
Wang(2023)은 프레임시프트 돌연변이 c.640_646del(p.R214Pfs*28)을 새로 동정하고, 완전 홍채결손·중심와형성부전·수정체 편위·망막박리를 보인 증례를 보고했습니다1).
Ratna 등(2022)은 인도 가계에서 런온 돌연변이 c.1268A>T(p.*423L)을 동정했습니다. 환자는 완전 무홍채·안진·중심와형성부전·AAK·수정체 상방 아탈구·고도 근시·시신경 위축을 보였으며, C-말단 연장 돌연변이에 의한 중증 표현형을 나타냈습니다6).
산발성 무홍채증에서는 PAX6에 더해 WT1 유전자를 포함한 대규모 결실이 WAGR 증후군의 원인이 됩니다. WT1 결실이 있는 경우 Wilms 종양 위험은 최대 50%입니다1). WAGR 증후군이 의심되면 유전학적 검사로 PAX6와 WT1의 결손을 확인하고, Wilms 종양 위험 평가와 발달 지연의 경과 관찰이 가능합니다8). 유전자 검사를 통한 WT1 영역의 평가가 필수적이며, 산발성 고립례의 30%는 5세까지 Wilms 종양을 발병한다고 알려져 있습니다. WT1 유전자가 PAX6의 근처에 위치하기 때문에, 둘 모두가 결손되는 제11염색체 단완 결실(11p13 결실)에서 무홍채증에 Wilms 종양이 합병됩니다.
질레스피 증후군은 ITPR1 유전자의 이형접합 우성음성 돌연변이 또는 양쪽 대립유전자 돌연변이에 의해 발생합니다3). 지금까지 분자 진단이 확인된 증례는 37예로 보고되었으며, Gly2554 잔기가 핫스팟으로 알려져 있습니다3).
무홍채증의 진단 기준(2020년)에 따라 다음 기준으로 확정합니다7).
A. 증상
B. 검사 소견
C. 감별해야 할 질환
E. 유전자 검사: PAX6 유전자의 병적 변이 또는 11p13 영역의 결실
진단 범주7):
난치병 인정을 위한 중증도 분류는 다음과 같은 4단계로 규정되어 있습니다7).
| 중증도 | 정의 |
|---|---|
| Ⅰ도 | 한쪽 눈 이환, 반대쪽 눈 정상 |
| Ⅱ도 | 양안 이환, 좋은 쪽의 교정시력 0.3 이상 |
| Ⅲ도 | 양안 이환, 좋은 쪽의 교정시력 0.1 이상 0.3 미만 |
| Ⅳ도 | 양안 이환, 좋은 쪽의 교정시력 0.1 미만 |
Ⅰ~Ⅲ도라도 녹내장 등에 의한 시야 협착(Goldmann I/4 시표로 중심 잔존 시야 20도 이내)을 동반하는 경우 한 단계 위의 중증도로 전환됩니다. 중증도 Ⅲ도 이상이 의료비 지원 대상이 됩니다7).
세극등 현미경으로 홍채의 결손 또는 형성 부전을 확인하면 임상 진단은 쉽습니다. 각경 검사나 초음파 생체 현미경으로 잔존 홍채 조직을 평가합니다. 전방각의 발달 이상 유무도 확인합니다.
다음 안 합병증을 체계적으로 평가합니다.
무홍채증의 유전학적 평가에서 가장 중요한 목표는 PAX6 결실이 WT1 유전자까지 미치는지 확인하는 것이다1). 전엑솜분석이나 MLPA법을 통해 PAX6 및 WT1 영역의 변이·결실을 평가한다1)2).
산발성 무홍채증에서는 WT1 유전자 결실로 인한 Wilms 종양 위험 평가가 생명 예후에 직결된다1). 가족성이라도 표현형의 다양성이 있으므로 유전자 검사를 통한 확진과 유전 상담이 권장된다.
무홍채증에 대한 근본적 치료법은 존재하지 않는다. 잔존 시력을 최대한 활용하는 저시력 관리와 각 합병증에 대한 개별 치료가 관리의 중심이 된다8).
각막 실질 혼탁에 대한 각막 이식은 신중히 결정한다8).
각막 이식으로 단기적으로 시력 개선을 얻을 수 있는 경우가 있지만, 황반 저형성 등의 동반 질환으로 인해 개선은 제한적이다. 장기적으로는 녹내장 진행 및 이식편 기능 부전으로 시력 예후가 불량해진다.
각막 상피 줄기세포 결핍증에서는 수술적 치료를 고려한다8).
백내장 수술은 혼탁과 눈부심 정도를 고려하여 검토한다8).
백내장은 20세까지 5085%에서 발생한다. 혼탁과 눈부심 강도에 따라 수술을 계획한다. 수술 증례의 66100%에서 시력 개선이 보고되었으나, 다음 사항에 주의가 필요하다.
Zinn 소대가 취약하므로, 안내 렌즈 삽입은 신중한 적응이 필요합니다.
Hu 등(2024)은 중증 AAK를 동반한 선천성 무홍채증 2예에 대해 샹들리에 역조명 보조 하에 수정체 유화 흡인술을 시행했습니다. 각막 혼탁으로 인해 일반적인 수술 중 시각화가 어려웠지만, 후방 조명으로 수정체와 전낭의 명확한 시각화가 가능해졌고, 수술 후 3주에 교정 시력이 각각 20/200 및 20/1000으로 개선되었습니다4).
녹내장은 시기능 예후에 직결되므로 적극적으로 치료합니다8).
녹내장 발병 후에는 다음 5단계 알고리즘에 따라 관리합니다.
약물 요법: 베타 차단제, 교감 신경 자극제, 프로스타글란딘(PG) 관련 약물이 효과적입니다. 유아에 대한 브리모니딘(α-아드레날린 수용체 자극제)은 중추 신경 억제 위험이 있으므로 2세 미만에는 금기입니다. 각막 상피 장애가 우려되는 경우 방부제가 포함되지 않은 제제를 사용합니다.
유출로 재건술(전방각 절개술, 섬유주 절개술): 첫 수술로 권장됩니다16). 예방적 전방각 절개술의 보고도 있습니다. 단, 잔존 홍채가 섬유주를 덮는 경우에는 효과가 없을 수 있습니다.
여과 수술(섬유주 절제술): 소수 사례 및 중단기 보고에 그칩니다. 소아 눈에서는 성적이 좋지 않은 경향이 있으며, 수술 후 안구로는 약 25%에 이릅니다13). 수술 후 악성 녹내장의 보고도 있습니다.
녹내장 임플란트 수술(튜브 션트 수술): Baerveldt 및 Ahmed형 장치를 사용할 수 있습니다. 유수정체안에 튜브를 삽입할 때는 각막 중심 방향이 아닌 접선 방향으로 권장합니다. 좋은 안압 조절이 기대됩니다.
섬모체 응고술: 최후의 수단입니다. 섬모체 냉동 응고에서는 대부분이 안구로에 빠졌다는 보고가 있습니다. 섬모체 저형성이 있으므로 정상안보다 안구로 위험이 높습니다.
각 발육 이상이 배경에 있기 때문에 일반적인 개방각 녹내장과는 다른 접근이 필요합니다. 첫 번째로 유출로 재건술을 선택하고, 다음으로 튜브 션트 수술이 좋은 선택지가 됩니다. 브리모니딘은 2세 미만에 사용 금기이며, 항대사 약물 사용은 AAK를 악화시킬 수 있으므로 신중한 판단이 요구됩니다8).
저시력 관리는 조기에 도입합니다8).
굴절 교정이 기본이며, 근시 합병률은 64% 이상으로 알려져 있습니다.
눈부심 치료는 시 기능 발달과 삶의 질 유지를 위해 중요합니다8).
대부분의 환자는 일반 학급에 진학할 수 있지만, 확대 교과서, 태블릿 단말기, 독서대 등의 지원이 필요합니다. 약시 학급에 다니거나 맹학교·시각특별지원학교에서 육아 상담·교육 상담을 이용하는 것도 선택지가 됩니다.
2017년 4월부터 지정 난치병으로 인정되어 신체장애인 수첩을 취득하지 않은 경우에도 중증도 Ⅲ도 이상이면 의료비 지원·보장구 지급 대상이 됩니다7). 대상 보장구에는 교정 안경, 차광 안경, 콘택트렌즈(인공 홍채 포함), 약시 안경, 시각장애인 안전 지팡이, 의안이 포함됩니다.
PAX6는 14개의 엑손을 포함하는 22kb의 게놈 DNA에 걸쳐 있으며, 422개의 아미노산을 코딩합니다1). 두 개의 DNA 결합 도메인(페어드 도메인과 페어드형 호메오도메인)을 가지고 있으며, C말단의 PST(프롤린·세린·트레오닌이 풍부한) 도메인이 전사 활성화 인자로 기능합니다.
PAX6는 세포의 증식, 분화, 이동, 접착을 조절하며, 그 표적에는 PAX6 자체 외에도 수정체 크리스탈린과 각막 케라틴을 코딩하는 유전자가 포함됩니다. 성인의 망막, 수정체, 각막에서도 발현이 지속됩니다. PAX6 유전자는 태생기의 기관 분화를 관장하는 마스터 컨트롤 유전자 중 하나입니다.
대부분의 PAX6 돌연변이는 넌센스 돌연변이 의존적 mRNA 분해(NMD)를 통해 하플로불충분을 유발합니다1). 조기 종결 코돈(PTC)을 도입하는 돌연변이(넌센스 돌연변이, 프레임시프트 돌연변이, 대부분의 스플라이스 돌연변이)가 전형적인 무홍채증 표현형을 초래합니다.
한편, PTC가 최종 엑손 또는 마지막에서 두 번째 엑손의 말단 50bp 이내에 위치하는 경우 NMD를 회피하고 절단형 단백질이 번역되어 중증 표현형을 나타낼 수 있다1).
PAX6의 무의미 돌연변이 c.282C>A(p.Cys94*)와 21번 삼염색체증이 동일 환자에서 동반된 드문 증례가 보고되었다. PAX6 돌연변이는 de novo로 발생하였으며, 완전 양안 무홍채증, 선천성 녹내장, AAK, 중심와 형성부전을 나타냈다2).
명확한 유전자형-표현형 상관관계는 확립되지 않았지만, 몇 가지 경향이 알려져 있습니다1).
Grant와 Walton의 전방각경 검사 시리즈에서는 초기에 홍채 실질이 섬유주대 위로 전방으로 확장되어 유착성 부착을 형성하고, 점차 시트 모양이 되어 최종적으로 전방각 폐쇄에 이르는 것이 나타났습니다14). 이 메커니즘이 녹내장 발병의 주요 원인입니다. 병리학적으로는 홍채 뿌리를 남긴 평활근 결손과 전방각 발달 부전이 기초를 이룹니다.
AAK는 주로 윤부 줄기세포 결핍(LSCD)에 의해 유발되지만, 각막 상피의 비정상 분화, 부착 이상, 결막 세포 침윤, 눈물 생성 부족도 관여합니다. PAX6에 의해 조절되는 기질 금속단백분해효소 9(MMP-9)의 결핍이 피브린 축적과 염증 세포 침윤을 유발하고, 실질의 콜라겐 배열이 흐트러져 투명성이 상실됩니다.
AAK는 5단계로 분류됩니다. Stage I에서는 주변부 상피만 비정상, Stage II에서는 구심성 상피 변화(중심부 미도달), Stage III에서는 중심 각막의 상피 변화와 주변부 표층 신생혈관, Stage IV에서는 전체 각막의 표층 신생혈관, Stage V에서는 전체 각막의 상피 이상과 심부 실질 반흔을 나타냅니다10).
PAX6 변이 상태와 AAK의 진행 사이에는 관련이 있습니다. PTC나 C-말단 연장 변이를 가진 환자에서는 AAK가 연령 의존적으로 진행되는 반면, 다른 변이형에서는 비진행성 각막증을 나타낼 수 있습니다11).
질레스피 증후군은 ITPR1 유전자의 변이로 인해 발생합니다3). ITPR1은 IP3 수용체 패밀리의 일원으로 Ca²⁺ 방출 채널을 형성하며 소포체에 국소화됩니다. 우성 음성 변이는 홍채 괄약근의 형성과 유지에 영향을 미쳐, 동공 주위의 특이적인 홍채 형성부전과 고정 산동을 초래합니다.
Ciaccio 등(2024)의 질레스피 증후군 문헌 검토에서는 분자 확인된 33예의 분석을 통해 운동 발달은 지연되지만 시간이 지남에 따라 개선되고, 지적 장애는 모든 예에서 나타나지 않으며 17%에서 정상 지능을 보이고, 신경학적 징후는 비진행성임이 확인되었습니다3).
전체 엑솜 분석 기술의 보급으로 새로운 PAX6 변이의 동정이 계속되고 있습니다. Human PAX6 Mutation Database에는 2018년 기준 491개의 변이가 등록되었으며, 이후 약 250개의 새로운 변이가 보고되었습니다1). 비코딩 영역의 변이가 무홍채증의 원인이 되는 사례도 확인되고 있어, 기존 검사로는 진단할 수 없었던 증례의 해명이 기대됩니다9).
중증 AAK를 동반한 증례에서의 백내장 수술에는 샹들리에 역조명 보조 시각화 기술이 유용합니다4). 이 기법은 Grade 3~4의 고도 AAK를 가진 환자에서도 안전한 수정체 유화 흡인술을 가능하게 하며, 수술 후 시력 개선이 얻어집니다.
PAX6 변이의 종류에 따라 AAK의 진행 패턴이 다른 것이 밝혀지고 있습니다. 유전자 검사의 비용 하락으로 변이형에 기반한 임상 경과 예측과 조기 개입이 현실적인 선택지가 되고 있습니다.
무홍채증과 21삼염색체증의 합병 증례에서는 두 질환의 공존에도 불구하고 비교적 경증의 경과를 보인 예가 보고되었습니다2). 여러 유전적 장애가 동일 환자에 공존할 때 표현형에 미치는 영향을 이해하는 것은 향후 개인 맞춤 의학에 중요한 지식을 제공할 가능성이 있습니다.
PTC형 변이에 대한 리드스루 약물(ataluren)의 무홍채증 적용이 기초 연구 수준에서 검토되고 있습니다8). PAX6 유전자 치료에 대해서는 Sey 변이 마우스 모델을 이용한 AAV-PAX6 벡터에 의한 유전자 보충 기초 연구가 진행 중입니다. 향후 임상 시험으로의 전개가 기대됩니다.
iPS 세포 유래 각막 상피 세포 시트 이식의 임상 시험이 국내외에서 실시되고 있으며, AAK에 대한 새로운 치료법으로 주목받고 있습니다8). 인공 홍채(CustomFlex Artificial Iris 등)는 해외에서 사용 실적이 축적되고 있습니다. 보조기구로서 인공 홍채가 부착된 콘택트렌즈는 보험 적용 대상입니다.
일본에서의 대규모 레지스트리 데이터 축적을 통한 실태 파악과 증거의 질 향상이 향후 중요한 과제입니다8). 개별 유전자 변이에 기반한 AAK의 진행 예측과 조기 개입의 최적화가 기대됩니다.