انعدام القزحية المعزول
التكرار: حوالي ثلثي الحالات.
نمط الوراثة: وراثة جسمية سائدة (AD).
الخصائص: ناتج عن طفرة في جين PAX6. لا يصاحبه أعراض جهازية. الاختراق كامل ولكن التعبيرية متغيرة.

انعدام القزحية (Aniridia) هو مرض خلقي نادر يتميز بنقص تنسج أو غياب القزحية بدرجات متفاوتة. اسم “انعدام القزحية” هو تسمية خاطئة، حيث يمكن دائمًا رؤية بقايا أنسجة القزحية باستخدام تنظير الزاوية (gonioscopy) أو المجهر الحيوي بالموجات فوق الصوتية (UBM).
يبلغ معدل الانتشار حوالي 1/40,000 إلى 1/100,000، ولا توجد فروق عرقية أو جنسية ملحوظة 1). يُصنف تحت رمز Q13.1 في التصنيف الدولي للأمراض (ICD-10).
انعدام القزحية هو مرض يصيب العين بأكملها، حيث لا يؤثر فقط على القزحية بل أيضًا على القرنية، العدسة، زاوية العين، النقرة، والعصب البصري 1)، ويسبب مضاعفات عينية متعددة تهدد الرؤية. يكون تشخيص الرؤية سيئًا بشكل عام، وغالبًا ما تبقى حدة البصر المصححة حوالي 0.1. يختفي رد فعل الحدقة ولكن يبقى رد فعل التكيف، وتكون الحالة ثنائية الجانب في 60-90% من الحالات.
يتم التعرف على الأنماط الظاهرية الثلاثة التالية.
انعدام القزحية المعزول
التكرار: حوالي ثلثي الحالات.
نمط الوراثة: وراثة جسمية سائدة (AD).
الخصائص: ناتج عن طفرة في جين PAX6. لا يصاحبه أعراض جهازية. الاختراق كامل ولكن التعبيرية متغيرة.
متلازمة WAGR
التكرار: جزء من الحالات المتفرقة.
نمط الوراثة: حذف متجاور لجيني PAX6 وWT1.
الخصائص: يرافقه ورم ويلمز، تشوهات الجهاز البولي التناسلي، والتخلف العقلي. خطر الورم يصل إلى 50%.
متلازمة غيليسبي
التكرار: حوالي 2% من الحالات.
نمط الوراثة: طفرة في جين ITPR1.
الخصائص: يرافقه رنح مخيخي وإعاقة ذهنية. يتميز بشذوذ قزحي محدد يتمثل في توسع حدقة ثابت 3).
يشكل انعدام القزحية المتفرق حوالي ثلث الحالات، وينتج عن حذف de novo في 11p13 يشمل PAX6. إذا امتد الحذف ليشمل جين WT1 المجاور، فإنه يسبب متلازمة WAGR 1). 25-30% من حالات انعدام القزحية المتفرق يصابون بورم ويلمز، ويبلغ الخطر النسبي 67.
PAX6 هو الجين الرئيسي المتحكم في تكوين العين، ويشارك في تطور العين، الأنبوب العصبي، البصلة الشمية، جزر لانغرهانس في البنكرياس، والظهارة الشمية. يحدث المرض بسبب فقدان وظيفة أليل واحد (نقص الجرعة)، بينما يؤدي شذوذ كلا الأليلين إلى وفاة الجنين. في عام 2017، تم إدراجه كمرض نادر محدد بموجب قانون الأمراض النادرة، ويصبح مؤهلاً للإعانة الطبية إذا كانت شدته من الدرجة الثالثة أو أعلى (انظر قسم التشخيص والفحص للحصول على التفاصيل) 7).
تمثل الحالات المتفرقة (الطفرات الجديدة) حوالي ثلث الحالات، ويمكن أن تحدث دون تاريخ عائلي. في الحالات المتفرقة، هناك احتمال لمتلازمة WAGR، لذلك من المهم إجراء الفحص الجيني وفحص الموجات فوق الصوتية للبطن لفحص ورم ويلمز.
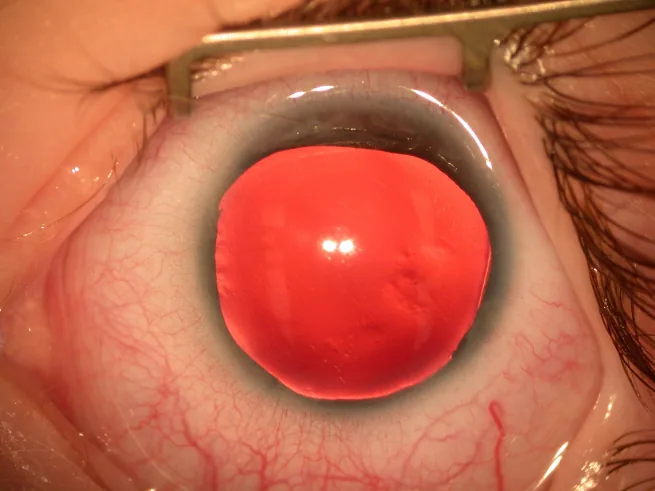
يتم اكتشاف معظم حالات انعدام القزحية عند الولادة بسبب شذوذ القزحية والحدقة، أو بسبب الرأرأة في مرحلة الرضاعة.
يختلف النمط الظاهري بين العائلات وداخل العائلة الواحدة، لكن الاختلاف بين العين اليمنى واليسرى عادة ما يكون صغيرًا.
بسبب نقص تنسج النقرة بشكل رئيسي، غالبًا ما تكون حدة البصر المصححة حوالي 0.1-0.2. يكون تشخيص البصر سيئًا بشكل خاص عند وجود نقص تنسج البقعة الصفراء. تصحيح الأخطاء الانكسارية ورعاية ضعف البصر منذ الطفولة المبكرة مهمان للنمو البصري.
نظرًا لأن PAX6 يُعبر عنه في أنسجة العين بالإضافة إلى الجهاز العصبي المركزي وجزر لانغرهانس في البنكرياس والظهارة الشمية، فقد تحدث المضاعفات خارج العين التالية 8).
العوامل المهمة التي تحدد الوظيفة البصرية هي الجلوكوما، نقص تنسج البقعة، الرأرأة، اعتلال القرنية، إعتام عدسة العين، وتشوهات القزحية. نظرًا لأن تلف المجال البصري وحدة البصر الناتج عن الجلوكوما لا رجعة فيه، فإن إدارة ضغط العين هي الأهم في المتابعة8).
معظم حالات انعدام القزحية الخلقي ناتجة عن طفرات متغايرة في جين PAX6 الموجود على الذراع القصير للكروموسوم 11 (11p13). آلية المرض الرئيسية هي نقص الجرعة (haploinsufficiency)1).
جين PAX6 هو جين تحكم رئيسي في تكوين العين، ويلعب دورًا مهمًا في تطور العين والأنبوب العصبي والبصيلة الشمية والبنكرياس. يتطلب التطور الطبيعي للعين نسختين من PAX6، وفقدان وظيفة نسخة واحدة فقط يؤدي إلى انعدام القزحية1).
في دراسة جماعية للمرضى الصينيين، تم تحديد طفرات مسببة في جين PAX6 في 96.9% من الحالات1). في انعدام القزحية النموذجي، يتم اكتشاف طفرات تحفز تحلل mRNA المعتمد على الطفرات غير المنطقية (NMD) أو حذف كبير في 96% من الحالات1).
من الناحية المرضية، هناك نقص في العضلات الملساء مع بقاء جذر القزحية، وتطور غير مكتمل لزاوية العين. يوجد خلل وظيفي في الخلايا الجذعية الظهارية للقرنية، مما يؤدي إلى تشوهات في الظهارة وغشاء بومان، وتكوين غشاء وعائي كثيف (pannus).
فيما يلي توزيع طفرات PAX6 المسببة للنمط الظاهري لانعدام القزحية.
| نوع الطفرة | التكرار |
|---|---|
| طفرة غير منطقية (nonsense) | حوالي 39% |
| طفرة إزاحة الإطار (frameshift) | حوالي 25% |
| طفرة في موقع الوصل (splice site) | حوالي 13% |
| طفرة مغلوطة | حوالي 12% |
تمثل الطفرات الانزلاقية (طفرات التمديد الطرفي C) حوالي 5%، حيث يتم إنتاج بروتين PAX6 ممتد بشكل غير طبيعي بسبب تحويل كودون التوقف إلى كودون ترجمة 6). غالبًا ما ترتبط طفرات التمديد الطرفي C بنقص تنسج القزحية الشديد وضعف البصر الشديد 1)6).
الطفرات الجينية غالبًا ما تكون من نوع PTC، كما توجد تقارير عن طفرات مغلوطة 7). فيما يتعلق بفائدة الفحص الجيني، يتم اكتشاف طفرات في حوالي 85% من حالات انعدام القزحية المعزول باستخدام تسلسل سانجر أو NGS. بالإضافة إلى ذلك، يتم اكتشاف حذف داخل جين PAX6 أو في المنطقة التنظيمية المحيطة به في حوالي 15% من الحالات باستخدام MLPA أو CMA 8).
حدد Wang (2023) طفرة إزاحة الإطار c.640_646del (p.R214Pfs*28) جديدة، وأبلغ عن حالة ظهرت فيها فقدان كامل للقزحية، ونقص تنسج النقرة، وانحراف العدسة، وانفصال الشبكية 1).
حدد Ratna وآخرون (2022) طفرة انزلاقية c.1268A>T (p.*423L) في عائلة هندية. أظهر المصابون انعدام قزحية كامل، ورأرأة، ونقص تنسج النقرة، وAAK، وخلع جزئي علوي للعدسة، وقصر نظر شديد، وضمور العصب البصري، مما يشير إلى نمط ظاهري شديد ناتج عن طفرة التمديد الطرفي C 6).
في حالات انعدام القزحية المتفرقة، يؤدي الحذف الكبير الذي يشمل جين WT1 بالإضافة إلى PAX6 إلى متلازمة WAGR. يصل خطر ورم ويلمز إلى 50% في حالة وجود حذف WT1 1). إذا كان هناك اشتباه في متلازمة WAGR، يمكن إجراء فحص جيني لتأكيد فقدان PAX6 وWT1، مما يتيح تقييم خطر ورم ويلمز ومتابعة تأخر النمو 8). يعد تقييم منطقة WT1 عن طريق الفحص الجيني أمرًا ضروريًا، حيث يُصاب 30% من الحالات المتفرقة بورم ويلمز قبل سن الخامسة. نظرًا لموقع جين WT1 بالقرب من PAX6، يؤدي حذف الذراع القصير للكروموسوم 11 (حذف 11p13) الذي يشمل كلا الجينين إلى حدوث انعدام القزحية مع ورم ويلمز.
تحدث متلازمة غيليسبي بسبب طفرات سلبية سائدة متغايرة أو طفرات ثنائية الأليل في جين ITPR1 3). تم الإبلاغ عن 37 حالة تم تأكيد تشخيصها جزيئيًا حتى الآن، ويُعرف بقايا Gly2554 كنقطة ساخنة 3).
بناءً على معايير تشخيص انعدام القزحية (2020)، يتم التأكيد وفقًا للمعايير التالية 7).
أ. الأعراض
ب. النتائج الفحصية
ج. الأمراض التي يجب تمييزها
هـ. الفحوصات الجينية: طفرة مرضية في جين PAX6 أو حذف في المنطقة 11p13
فئات التشخيص7):
يُحدد تصنيف الشدة لتأهيل الأمراض المستعصية بالمراحل الأربع التالية7).
| الشدة | التعريف |
|---|---|
| الدرجة الأولى | إصابة عين واحدة، والعين الأخرى سليمة |
| الدرجة الثانية | إصابة كلتا العينين، حدة الإبصر المصححة في العين الأفضل ≥ 0.3 |
| الدرجة الثالثة | إصابة كلتا العينين، حدة الإبصر المصححة في العين الأفضل ≥ 0.1 وأقل من 0.3 |
| الدرجة الرابعة | إصابة كلتا العينين، حدة الإبصر المصححة في العين الأفضل أقل من 0.1 |
حتى في الدرجات من الأولى إلى الثالثة، إذا كان هناك تضيق في المجال البصري بسبب الجلوكوما أو غيره (المجال البصري المركزي المتبقي ≤ 20 درجة باستخدام هدف Goldmann I/4)، تنتقل الحالة إلى درجة شدة أعلى. الدرجة الثالثة فما فوق مؤهلة للحصول على دعم التكاليف الطبية7).
التشخيص السريري سهل إذا تم تأكيد غياب القزحية أو عدم اكتمال تكوينها باستخدام المصباح الشقي. يتم تقييم الأنسجة القزحية المتبقية باستخدام تنظير زاوية العين أو المجهر فوق الصوتي الحيوي. كما يتم التحقق من وجود تشوهات في زاوية الغرفة الأمامية.
يتم تقييم المضاعفات العينية التالية بشكل منهجي:
الهدف الأهم في التقييم الجيني لانعدام القزحية هو التأكد مما إذا كان حذف PAX6 يمتد ليشمل جين WT11). يتم تقييم الطفرات والحذف في منطقتي PAX6 وWT1 باستخدام تسلسل الإكسوم الكامل أو تقنية MLPA1)2).
في حالات انعدام القزحية المتفرقة، يرتبط تقييم خطر ورم ويلمز الناتج عن حذف جين WT1 بشكل مباشر بالبقاء على قيد الحياة1). حتى في الحالات العائلية، نظرًا لتنوع النمط الظاهري، يُوصى بالتشخيص المؤكد عبر الاختبار الجيني والاستشارة الوراثية.
لا يوجد علاج جذري لانعدام القزحية. يركز العلاج على رعاية ضعف البصر لتعظيم الاستفادة من الرؤية المتبقية، والعلاج الفردي لكل مضاعفة8).
يجب توخي الحذر عند إجراء زراعة القرنية لعتمة حمة القرنية8).
قد تؤدي زراعة القرنية إلى تحسن مؤقت في الرؤية، لكن التحسن محدود بسبب مضاعفات مثل نقص تنسج البقعة. على المدى الطويل، يكون تشخيص الرؤية سيئًا بسبب تقدم الجلوكوما وفشل الطعم.
في حالة استنزاف الخلايا الجذعية الظهارية القرنية، يُنظر في العلاج الجراحي 8).
يتم النظر في جراحة الساد بناءً على درجة العتامة ورهاب الضوء 8).
يحدث الساد في 50-85% من الحالات قبل سن 20 عامًا. يتم التخطيط للجراحة بناءً على شدة العتامة ورهاب الضوء. يُذكر أن 66-100% من الحالات الجراحية شهدت تحسنًا في الرؤية، ولكن يجب الانتباه إلى النقاط التالية.
نظرًا لضعف الألياف الناحلة (Zinn)، فإن إدخال العدسة داخل العين يتطلب مؤشرات حذرة.
أجرى هو وزملاؤه (2024) عملية استحلاب العدسة بمساعدة الإضاءة الخلفية الثريا لحالتين من انعدام القزحية الخلقي المصحوب باعتام قرنية شديد. على الرغم من صعوبة الرؤية الجراحية المعتادة بسبب عتامة القرنية، إلا أن الإضاءة من الخلف أتاحت رؤية واضحة للعدسة والمحفظة الأمامية، وتحسنت حدة البصر المصححة إلى 20/200 و20/1000 بعد 3 أسابيع من الجراحة 4).
يجب علاج الجلوكوما بنشاط لأنها تؤثر بشكل مباشر على النتائج البصرية 8).
بعد ظهور الجلوكوما، تتم الإدارة وفقًا للخوارزمية المكونة من 5 خطوات التالية.
العلاج الدوائي: حاصرات بيتا، محفزات الودي، ومضاهئات البروستاجلاندين فعالة. يُمنع استخدام بريمونيدين (محفز مستقبلات ألفا الأدرينالية) للأطفال دون سن الثانية بسبب خطر تثبيط الجهاز العصبي المركزي. في حالة القلق من تلف ظهارة القرنية، استخدم مستحضرات خالية من المواد الحافظة.
إعادة بناء مسار التصريف (بضع الزاوية أو بضع التربيق): يوصى به كجراحة أولى 16). هناك تقارير عن بضع الزاوية الوقائي. ومع ذلك، قد يكون غير فعال في الحالات التي تغطي فيها القزحية المتبقية التربيق.
جراحة الترشيح (استئصال التربيق): تقتصر التقارير على حالات قليلة ومتوسطة المدى. تميل النتائج إلى أن تكون ضعيفة في عيون الأطفال، ويصل تسرب العين بعد الجراحة إلى حوالي 25% 13). هناك أيضًا تقارير عن الجلوكوما الخبيثة بعد الجراحة.
جراحة زرع تحويلة الجلوكوما (جراحة التحويلة الأنبوبية): تتوفر أجهزة من نوع Baerveldt وAhmed. يُوصى بإدخال الأنبوب في العيون التي تحتوي على عدسة طبيعية بشكل مماسي وليس نحو مركز القرنية. يمكن توقع تحكم جيد في ضغط العين.
تخثير الجسم الهدبي: الملاذ الأخير. هناك تقارير عن أن التجميد بالتبريد للجسم الهدبي أدى إلى تسرب العين في العديد من الحالات. نظرًا لوجود نقص تنسج في الجسم الهدبي، فإن خطر تسرب العين أعلى من العين السليمة.
نظرًا لوجود تشوهات في زاوية الغرفة الأمامية، يلزم اتباع نهج مختلف عن الجلوكوما مفتوحة الزاوية العادية. في البداية، يُختار إعادة بناء مسار التصريف، ثم جراحة تحويلة الأنبوب كخيار جيد. يُمنع استخدام بريمونيدين للأطفال دون سن الثانية، ويتطلب استخدام مضادات الأيض تقييمًا دقيقًا لأنها قد تؤدي إلى تفاقم AAK 8).
يجب تقديم رعاية ضعف البصر في مرحلة مبكرة 8).
تصحيح الانكسار هو الأساس، ويُقدر معدل الإصابة بقصر النظر بأكثر من 64%.
علاج رهاب الضوء مهم للحفاظ على تطور الوظيفة البصرية وجودة الحياة 8).
يمكن لمعظم المرضى الالتحاق بالمدارس العادية، لكنهم يحتاجون إلى دعم مثل الكتب المدرسية الموسعة والأجهزة اللوحية وحوامل القراءة. كما أن الالتحاق بفصول ضعاف البصر أو الاستفادة من استشارات التربية والتعليم في مدارس المكفوفين ومدارس الدعم البصري الخاص هي خيارات متاحة.
منذ أبريل 2017، تم تصنيف هذا المرض كمرض نادر محدد، وبالتالي حتى إذا لم يحصل المريض على بطاقة إعاقة بصرية، فإنه مؤهل للحصول على دعم النفقات الطبية وتوفير الأجهزة التعويضية إذا كانت شدته من الدرجة الثالثة أو أعلى 7). تشمل الأجهزة التعويضية المؤهلة النظارات الطبية، النظارات الواقية من الضوء، العدسات اللاصقة (بما في ذلك تلك المزودة بقزحية اصطناعية)، نظارات ضعاف البصر، عصا الأمان للمكفوفين، والعين الاصطناعية.
يمتد جين PAX6 على 22 كيلو قاعدة من الحمض النووي الجينومي ويحتوي على 14 إكسونًا، ويشفر 422 حمضًا أمينيًا 1). يحتوي على نطاقين رابطين للحمض النووي (النطاق المزدوج والنطاق المنزلي المزدوج)، ويعمل نطاق PST (الغني بالبرولين والسيرين والثريونين) في الطرف الكربوكسيلي كمنشط للنسخ.
ينظم PAX6 تكاثر الخلايا وتمايزها وهجرتها والتصاقها، وتشمل أهدافه جين PAX6 نفسه بالإضافة إلى الجينات المشفرة للكريستالين البلوري والكيراتين القرني. يستمر التعبير عنه في شبكية العين والعدسة والقرنية لدى البالغين. جين PAX6 هو أحد الجينات الرئيسية المنظمة لتمايز الأعضاء خلال المرحلة الجنينية.
تسبب معظم طفرات PAX6 قصورًا فردانيًا عبر تحلل الحمض النووي الريبوزي الرسول المعتمد على الطفرات غير المعنوية (NMD) 1). تؤدي الطفرات التي تُدخل كودون توقف مبكر (PTC) (الطفرات غير المعنوية، طفرات تغيير الإطار، ومعظم طفرات التوصيل) إلى النمط الظاهري النموذجي لانعدام القزحية.
من ناحية أخرى، إذا كان PTC يقع في الإكسون الأخير أو ضمن آخر 50 زوجًا قاعديًا من الإكسون قبل الأخير، فإنه يفلت من NMD، ويتم ترجمة بروتين مبتور مما قد يؤدي إلى نمط ظاهري شديد 1).
تم الإبلاغ عن حالة نادرة لطفرة غير معنوية في PAX6 c.282C>A (p.Cys94*) مصاحبة لتثلث الصبغي 21 لدى نفس المريض. حدثت طفرة PAX6 بشكل جديد، وأظهرت انعدام القزحية الكامل الثنائي، الجلوكوما الخلقية، AAK، ونقص تنسج النقرة 2).
على الرغم من عدم وجود ارتباط واضح بين النمط الجيني والنمط الظاهري، إلا أن بعض الاتجاهات معروفة 1).
في سلسلة تنظير الزاوية لجرانت ووالتون، تبين أن سدى القزحية يمتد أماميًا فوق الشبكة التربيقية مكونًا التصاقات تشبه الالتصاق، ثم يصبح صفائحيًا تدريجيًا حتى يؤدي إلى انسداد الزاوية 14). هذه الآلية هي العامل الرئيسي في تطور الجلوكوما. من الناحية المرضية، يكمن الأساس في نقص العضلات الملساء مع بقاء جذر القزحية ونقص تطور الزاوية.
يحدث اعتلال القرنية القزحي (AAK) بشكل رئيسي بسبب نقص الخلايا الجذعية للحوف (LSCD)، ولكن يشارك أيضًا التمايز غير الطبيعي للظهارة القرنية، واضطراب الالتصاق، وارتشاح الخلايا الملتحمةية، ونقص إنتاج الدموع. يؤدي نقص المصفوفة المعدنية البروتياز 9 (MMP-9) المنظمة بواسطة PAX6 إلى تراكم الفيبرين وارتشاح الخلايا الالتهابية، مما يسبب اضطراب ترتيب الكولاجين في السدى وفقدان الشفافية.
يصنف اعتلال القرنية القزحي (AAK) إلى 5 مراحل. في المرحلة الأولى، يكون الشذوذ في الظهارة المحيطية فقط، وفي المرحلة الثانية، تغيرات ظهارية جاذبة للمركز (لم تصل إلى المركز)، وفي المرحلة الثالثة، تغيرات ظهارية في مركز القرنية وأوعية دموية سطحية محيطية، وفي المرحلة الرابعة، أوعية دموية سطحية في القرنية بأكملها، وفي المرحلة الخامسة، شذوذ ظهاري في القرنية بأكملها وتندب في السدى العميق 10).
هناك علاقة بين حالة طفرة PAX6 وتطور اعتلال القرنية القزحي (AAK). في المرضى الذين لديهم طفرات PTC أو استطالة طرفية C، يتطور AAK بشكل يعتمد على العمر، بينما في الأنماط الطفرية الأخرى، قد يظهر اعتلال قرنية غير تقدمي 11).
تحدث متلازمة غيليسبي بسبب طفرات في جين ITPR1 3). ITPR1 هو عضو في عائلة مستقبلات IP3 التي تشكل قنوات إطلاق الكالسيوم Ca²⁺ وتتمركز في الشبكة الإندوبلازمية. تؤثر الطفرات السلبية السائدة على تكوين وصيانة العضلة العاصرة للقزحية، مما يؤدي إلى نقص تنسج قزحية محدد حول الحدقة وتوسع حدقة ثابت.
في مراجعة أدبيات متلازمة غيليسبي التي أجراها تشاتشو وآخرون (2024)، من تحليل 33 حالة مؤكدة جزيئيًا، تأكد أن التطور الحركي يتأخر ولكنه يتحسن بمرور الوقت، وأن الإعاقة الذهنية لا تحدث في جميع الحالات حيث أن 17% لديهم ذكاء طبيعي، وأن العلامات العصبية غير تقدمية 3).
مع انتشار تقنية تحليل الإكسوم الكامل، يستمر تحديد طفرات PAX6 الجديدة. اعتبارًا من عام 2018، تم تسجيل 491 طفرة في قاعدة بيانات طفرات PAX6 البشرية، وتم الإبلاغ عن حوالي 250 طفرة جديدة منذ ذلك الحين1). كما يتم تحديد طفرات في المناطق غير المشفرة كسبب لانعدام القزحية، مما قد يساعد في تشخيص الحالات التي لم يتم تشخيصها سابقًا9).
في حالات إعتام عدسة العين المصحوب باعتلال القرنية الشديد المرتبط بانعدام القزحية، تكون تقنية التصور بمساعدة الإضاءة الخلفية الثريا مفيدة4). تتيح هذه التقنية إجراء عملية استحلاب العدسة بأمان حتى في المرضى الذين يعانون من اعتلال القرنية الشديد من الدرجة 3-4، مما يحسن الرؤية بعد الجراحة.
يتضح أن أنواع طفرات PAX6 تؤدي إلى أنماط مختلفة من تطور اعتلال القرنية المرتبط بانعدام القزحية. مع انخفاض تكلفة الاختبارات الجينية، أصبح التنبؤ بالمسار السريري بناءً على نوع الطفرة والتدخل المبكر خيارًا عمليًا.
في حالات انعدام القزحية المصحوب بمتلازمة داون، تم الإبلاغ عن مسار سريري خفيف نسبيًا على الرغم من وجود المرضين معًا2). فهم تأثير الاضطرابات الجينية المتعددة على النمط الظاهري لدى نفس المريض قد يوفر رؤى مهمة للطب الشخصي في المستقبل.
يتم دراسة تطبيق دواء أتالورين (الذي يتجاوز طفرات نوع PTC) في انعدام القزحية على المستوى الأساسي8). بالنسبة للعلاج الجيني لـ PAX6، تجري أبحاث أساسية على استبدال الجين باستخدام ناقل AAV-PAX6 في نموذج فأر بطفرات Sey. من المتوقع أن تتطور هذه الأبحاث إلى تجارب سريرية في المستقبل.
تُجرى تجارب سريرية لزرع صفائح الخلايا الظهارية القرنية المشتقة من الخلايا الجذعية المحفزة في الداخل والخارج، مما يجعلها علاجًا واعدًا لاعتلال القرنية المرتبط بانعدام القزحية8). بالنسبة للقزحية الاصطناعية (مثل CustomFlex Artificial Iris)، تراكمت خبرة الاستخدام في الخارج. العدسات اللاصقة المزودة بقزحية اصطناعية كأجهزة تعويضية مشمولة بالتأمين.
يعد تجميع بيانات السجلات واسعة النطاق في اليابان لفهم الوضع الفعلي وتحسين جودة الأدلة من القضايا المهمة في المستقبل8). من المتوقع تحسين التنبؤ بتطور اعتلال القرنية المرتبط بانعدام القزحية بناءً على الطفرات الجينية الفردية وتحسين التدخل المبكر.