Изолированная аниридия
Частота: около 2/3 всех случаев.
Тип наследования: аутосомно-доминантный (АД).
Особенности: обусловлена мутациями гена PAX6. Не сопровождается системными проявлениями. Пенетрантность полная, но экспрессивность вариабельна.

Аниридия — редкое врожденное заболевание, характеризующееся различной степенью недоразвития или отсутствия радужной оболочки. Название «аниридия» является неточным, так как при гониоскопии или ультразвуковой биомикроскопии (УБМ) почти всегда обнаруживаются фрагменты ткани радужки.
Распространенность составляет примерно 1:40 000–1:100 000; значимых расовых или половых различий не описано 1). По МКБ-10 классифицируется как Q13.1.
Это панокулярное заболевание, поражающее не только радужку, но и роговицу, хрусталик, угол передней камеры, центральную ямку и зрительный нерв 1), и проявляющееся множеством угрожающих зрению глазных осложнений. Прогноз по зрению в целом неблагоприятный; корригированная острота зрения часто составляет около 0,1. Зрачковая реакция отсутствует, но аккомодация сохранена; в 60–90% случаев поражение двустороннее.
Выделяют следующие три фенотипа.
Изолированная аниридия
Частота: около 2/3 всех случаев.
Тип наследования: аутосомно-доминантный (АД).
Особенности: обусловлена мутациями гена PAX6. Не сопровождается системными проявлениями. Пенетрантность полная, но экспрессивность вариабельна.
Синдром WAGR
Частота: часть спорадических случаев.
Тип наследования: смежная делеция генов PAX6 и WT1.
Особенности: сочетается с опухолью Вильмса, аномалиями мочеполовой системы и умственной отсталостью. Риск опухоли достигает 50%.
Синдром Жиляспи
Частота: около 2% всех случаев.
Тип наследования: мутации гена ITPR1.
Особенности: сопровождается мозжечковой атаксией и интеллектуальными нарушениями. Характерна специфическая аномалия радужки с фиксированным мидриазом3).
Спорадическая аниридия составляет около 1/3 всех случаев и возникает вследствие de novo делеции в 11p13, включающей PAX6. Если делеция распространяется на соседний ген WT1, это приводит к синдрому WAGR1). У 25–30% пациентов со спорадической аниридией развивается опухоль Вильмса, относительный риск составляет 67.
PAX6 является главным регуляторным геном формирования глаза и участвует в развитии глаза, нервной трубки, обонятельной луковицы, островков Лангерганса поджелудочной железы и обонятельного эпителия. Заболевание возникает при потере функции одной аллели (гаплонедостаточность), а мутации в обеих аллелях приводят к летальному исходу на эмбриональной стадии. В 2017 году аниридия была включена в перечень редких заболеваний, определяемых Законом о редких заболеваниях, и при тяжести III степени и выше (см. раздел «Диагностика и обследование») предоставляется финансовая поддержка на медицинские расходы7).
Спорадические случаи (новые мутации) составляют около 1/3 всех случаев и могут возникать без семейного анамнеза. При спорадических случаях существует вероятность синдрома WAGR, поэтому важны генетическое тестирование и скрининг на опухоль Вильмса с помощью УЗИ брюшной полости.
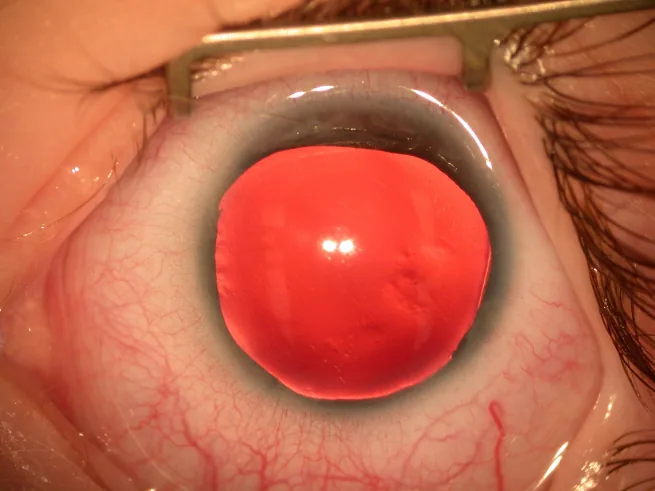
Большинство случаев анниридии выявляется при рождении из-за аномалий радужки и зрачка или в младенчестве из-за нистагма.
Фенотип варьирует между семьями и внутри семьи, но различия между правым и левым глазом обычно невелики.
Основной причиной является гипоплазия фовеа, поэтому корригированная острота зрения часто составляет 0,1–0,2. При сочетании с гипоплазией макулы прогноз зрения особенно неблагоприятный. Коррекция рефракции и низкозрительная помощь с раннего детства важны для зрительного развития.
PAX6 экспрессируется не только в тканях глаза, но и в центральной нервной системе, островках Лангерганса поджелудочной железы и обонятельном эпителии, поэтому могут наблюдаться следующие внеглазные осложнения 8).
Важными факторами, определяющими зрительную функцию, являются глаукома, гипоплазия желтого пятна, нистагм, кератопатия, катаракта и аномалии радужки. Поскольку нарушения поля зрения и остроты зрения, вызванные глаукомой, необратимы, контроль внутриглазного давления является наиболее важным при наблюдении8).
Большинство случаев врожденной аниридии вызваны гетерозиготными мутациями в гене PAX6, расположенном на коротком плече 11-й хромосомы (11p13). Основным механизмом развития является гаплонедостаточность PAX61).
Ген PAX6 является главным регуляторным геном формирования глаза и играет важную роль в развитии глаза, нервной трубки, обонятельной луковицы и поджелудочной железы. Для нормального развития глаза необходимы две копии PAX6, и потеря функции одной копии приводит к аниридии1).
В когортном исследовании китайских пациентов причинные мутации в гене PAX6 были выявлены в 96,9% случаев1). При типичной аниридии в 96% случаев обнаруживаются мутации, индуцирующие нонсенс-опосредованный распад мРНК (NMD), или крупные делеции1).
Патологически наблюдается отсутствие гладких мышц при сохранении корня радужки, а также недоразвитие угла передней камеры. Отмечается дисфункция стволовых клеток эпителия роговицы, приводящая к аномалиям эпителия и мембраны Боумена, а также к образованию васкуляризированного паннуса.
Ниже приведено распределение мутаций PAX6, вызывающих фенотип аниридии.
| Тип мутации | Частота |
|---|---|
| Нонсенс-мутация | ~39% |
| Мутация сдвига рамки | ~25% |
| Сплайс-мутация | ~13% |
| миссенс-мутация | около 12% |
Мутации с удлинением (C-концевые мутации) составляют около 5% и приводят к продукции аномально удлиненного белка PAX6 из-за преобразования стоп-кодона в кодирующий кодон 6). C-концевые мутации часто сопровождаются тяжелой гипоплазией радужки и выраженными нарушениями зрения 1)6).
Генетические мутации чаще всего относятся к PTC-типу, также описаны миссенс-мутации 7). Что касается полезности генетического тестирования, с помощью секвенирования по Сэнгеру или NGS мутации выявляются почти у 85% пациентов с изолированной аниридией. Кроме того, с помощью MLPA или CMA делеции в гене PAX6 или цис-регуляторной области обнаруживаются почти у 15% пациентов 8).
Wang (2023) впервые идентифицировал мутацию со сдвигом рамки c.640_646del (p.R214Pfs*28) и сообщил о случае с полным отсутствием радужки, гипоплазией фовеа, эктопией хрусталика и отслойкой сетчатки 1).
Ratna и соавт. (2022) идентифицировали мутацию с удлинением c.1268A>T (p.*423L) в индийской семье. У пораженных наблюдались полная аниридия, нистагм, гипоплазия фовеа, ААК, верхний подвывих хрусталика, миопия высокой степени и атрофия зрительного нерва, что указывает на тяжелый фенотип, вызванный C-концевой мутацией 6).
При спорадической аниридии крупные делеции, включающие ген WT1 в дополнение к PAX6, вызывают синдром WAGR. Риск опухоли Вильмса при делеции WT1 составляет до 50% 1). При подозрении на синдром WAGR генетическое тестирование позволяет подтвердить делеции PAX6 и WT1, оценить риск опухоли Вильмса и наблюдать за задержкой развития 8). Оценка области WT1 с помощью генетического тестирования необходима; считается, что у 30% спорадических случаев опухоль Вильмса развивается к 5 годам. Поскольку ген WT1 расположен рядом с PAX6, делеция короткого плеча хромосомы 11 (11p13), затрагивающая оба гена, приводит к сочетанию аниридии с опухолью Вильмса.
Синдром Жиляспи вызывается гетерозиготной доминантно-негативной или биаллельной мутацией гена ITPR1 3). Сообщается о 37 подтвержденных молекулярной диагностикой случаях, причем остаток Gly2554 известен как горячая точка 3).
Диагноз аниридии устанавливается на основании следующих критериев (2020 г.)7).
A. Симптомы
B. Данные обследования
C. Заболевания для дифференциальной диагностики
E. Генетическое тестирование: патогенная мутация гена PAX6 или делеция в области 11p13
Диагностические категории7):
Классификация по степени тяжести для признания заболевания редким (нанбё) определена в четырех стадиях7).
| Степень тяжести | Определение |
|---|---|
| I степень | Поражение одного глаза, другой глаз здоров |
| II степень | Поражение обоих глаз, корригированная острота зрения лучшего глаза 0,3 и выше |
| III степень | Поражение обоих глаз, корригированная острота зрения лучшего глаза от 0,1 до 0,3 |
| IV степень | Поражение обоих глаз, корригированная острота зрения лучшего глаза менее 0,1 |
Даже при I–III степени, если имеется сужение поля зрения вследствие глаукомы и т.п. (центральное остаточное поле зрения в пределах 20° по Goldmann I/4), степень тяжести повышается на одну ступень. Степень тяжести III и выше является основанием для субсидирования медицинских расходов 7).
Клиническая диагностика проста при выявлении дефекта или недоразвития радужки с помощью щелевой лампы. Оценка остаточной ткани радужки проводится с помощью гониоскопии или ультразвуковой биомикроскопии. Также проверяется наличие аномалий развития угла передней камеры.
Систематически оцениваются следующие глазные осложнения:
Наиболее важной целью генетической оценки аниридии является подтверждение того, распространяется ли делеция PAX6 на ген WT11). Полноэкзомное секвенирование и MLPA используются для оценки мутаций и делеций в областях PAX6 и WT11)2).
При спорадической аниридии оценка риска опухоли Вильмса из-за делеции гена WT1 напрямую влияет на прогноз выживаемости1). Даже при семейных случаях из-за фенотипической вариабельности рекомендуется подтверждение диагноза с помощью генетического тестирования и генетическое консультирование.
Радикального лечения аниридии не существует. Основой ведения является слабовидение, максимально использующее остаточное зрение, и индивидуальное лечение каждого осложнения8).
Трансплантация роговицы при помутнении стромы роговицы должна проводиться с осторожностью8).
Трансплантация роговицы может краткосрочно улучшить зрение, но улучшение ограничено из-за сопутствующих состояний, таких как гипоплазия макулы. В долгосрочной перспективе прогрессирование глаукомы и дисфункция трансплантата приводят к плохому зрительному прогнозу.
При недостаточности лимбальных стволовых клеток роговицы рассматривается хирургическое лечение 8).
Хирургия катаракты рассматривается с учетом степени помутнения и светобоязни 8).
Катаракта развивается у 50–85% пациентов к 20 годам. Операция планируется в зависимости от интенсивности помутнения и светобоязни. Сообщается, что у 66–100% прооперированных пациентов наблюдается улучшение зрения, однако необходимо учитывать следующие моменты.
Из-за слабости цинновых связок имплантация интраокулярной линзы требует осторожного подбора показаний.
Hu и соавт. (2024) выполнили факоэмульсификацию с использованием хрусталика с задним освещением у двух пациентов с врожденной аниридией и тяжелой ААК. Из-за помутнения роговицы обычная интраоперационная визуализация была затруднена, но освещение сзади позволило четко визуализировать хрусталик и переднюю капсулу; через 3 недели после операции корригированная острота зрения улучшилась до 20/200 и 20/1000 соответственно 4).
Глаукома напрямую влияет на зрительный прогноз, поэтому ее следует активно лечить 8).
После развития глаукомы ведение осуществляется по следующему 5-этапному алгоритму.
Медикаментозная терапия: эффективны бета-блокаторы, симпатомиметики и препараты простагландинового ряда. Бримонидин (альфа-адреномиметик) противопоказан детям младше 2 лет из-за риска угнетения ЦНС. При подозрении на повреждение эпителия роговицы следует использовать препараты без консервантов.
Восстановление путей оттока (гониотомия, трабекулотомия): рекомендуется в качестве первой операции 16). Имеются сообщения о профилактической гониотомии. Однако в случаях, когда остаточная радужка покрывает трабекулярную сеть, процедура может быть неэффективной.
Фильтрующая хирургия (трабекулэктомия): имеются лишь краткосрочные и среднесрочные сообщения на небольшом числе пациентов. У детей результаты, как правило, хуже; послеоперационная гипотония глазного яблока возникает примерно в 25% случаев 13). Также сообщалось о послеоперационной злокачественной глаукоме.
Имплантация глаукомного дренажа (трубчатый шунт): доступны устройства типа Baerveldt и Ahmed. При имплантации трубки в факичный глаз рекомендуется направлять ее не к центру роговицы, а по касательной. Ожидается хороший контроль внутриглазного давления.
Циклокоагуляция: метод последнего выбора. Сообщается, что при криокоагуляции цилиарного тела у многих пациентов развивается гипотония глазного яблока. Из-за гипоплазии цилиарного тела риск гипотонии выше, чем в здоровых глазах.
Из-за аномалий развития угла передней камеры требуется иной подход, чем при обычной открытоугольной глаукоме. Первым выбором является реконструкция путей оттока, затем хорошим вариантом становится шунтирующая хирургия. Бримонидин противопоказан детям младше 2 лет, а применение антиметаболитов может усугубить ААК, поэтому требуется осторожное решение 8).
Уход при слабовидении следует начинать на ранних стадиях8).
Основой является коррекция рефракции, частота сопутствующей миопии составляет 64% и более.
Лечение светобоязни важно для сохранения развития зрительных функций и качества жизни8).
Большинство пациентов могут посещать обычные классы, но им может потребоваться поддержка, такая как увеличенные учебники, планшеты и подставки для книг. Также возможны варианты посещения классов для слабовидящих или консультации по воспитанию и образованию в школах для слепых и специализированных школах для детей с нарушениями зрения.
С апреля 2017 года это заболевание признано редким, поэтому даже без получения удостоверения инвалидности пациенты с тяжестью III степени и выше имеют право на субсидирование медицинских расходов и предоставление вспомогательных средств7). К вспомогательным средствам относятся корректирующие очки, светозащитные очки, контактные линзы (включая с искусственной радужкой), очки для слабовидящих, трость для слепых и глазной протез.
Ген PAX6 охватывает 22 кб геномной ДНК, включая 14 экзонов, и кодирует 422 аминокислоты1). Он содержит два ДНК-связывающих домена (парный домен и парный гомеодомен), а C-концевой PST-домен (богатый пролином, серином и треонином) функционирует как активатор транскрипции.
PAX6 регулирует пролиферацию, дифференцировку, миграцию и адгезию клеток, а его мишени включают сам PAX6, а также гены, кодирующие кристаллины хрусталика и кератины роговицы. Экспрессия PAX6 сохраняется в сетчатке, хрусталике и роговице взрослых. Ген PAX6 является одним из мастер-контрольных генов, управляющих органогенезом в эмбриональном периоде.
Большинство мутаций PAX6 вызывают гаплонедостаточность через нонсенс-опосредованный распад мРНК (NMD)1). Мутации, вводящие преждевременный стоп-кодон (PTC) (нонсенс-мутации, мутации сдвига рамки и большинство сплайсинговых мутаций), приводят к типичному фенотипу аниридии.
С другой стороны, если PTC расположен в последнем экзоне или в пределах 50 п.н. от конца предпоследнего экзона, он может избежать NMD, что приводит к трансляции усеченного белка и потенциально тяжелому фенотипу1).
Сообщалось о редком случае сочетания нонсенс-мутации PAX6 c.282C>A (p.Cys94*) и трисомии 21 у одного пациента. Мутация PAX6 возникла de novo и привела к полной двусторонней аниридии, врожденной глаукоме, ААК и гипоплазии фовеа2).
Хотя четкая генотип-фенотипическая корреляция не установлена, известны некоторые тенденции1).
В серии гониоскопических исследований Grant и Walton было показано, что на ранних стадиях строма радужки распространяется кпереди на трабекулярную сеть, образуя синехиеподобные сращения, которые постепенно становятся листовидными и в конечном итоге приводят к закрытию угла14). Этот механизм является основным в развитии глаукомы. Патологически в основе лежит дефект гладкой мышцы с сохранением корня радужки и недоразвитие угла передней камеры.
ААК в основном вызывается недостаточностью лимбальных стволовых клеток (LSCD), однако также участвуют аномальная дифференцировка и адгезия эпителия роговицы, инфильтрация конъюнктивальных клеток и недостаточная продукция слезной жидкости. Дефицит матриксной металлопротеиназы 9 (MMP-9), регулируемой PAX6, приводит к накоплению фибрина и инфильтрации воспалительных клеток, а нарушение коллагеновой архитектуры стромы ведет к потере прозрачности.
ААК классифицируется на 5 стадий. Стадия I: изменения только периферического эпителия; Стадия II: центростремительные эпителиальные изменения (центр не затронут); Стадия III: эпителиальные изменения центральной роговицы и поверхностная неоваскуляризация по периферии; Стадия IV: поверхностная неоваскуляризация всей роговицы; Стадия V: эпителиальные изменения всей роговицы и глубокие стромальные рубцы10).
Существует связь между статусом мутации PAX6 и прогрессированием ААК. У пациентов с PTC или мутациями, удлиняющими C-конец, ААК прогрессирует с возрастом, тогда как при других типах мутаций может наблюдаться непрогрессирующая кератопатия11).
Синдром Жиляспи вызывается мутациями в гене ITPR13). ITPR1 является членом семейства рецепторов IP3, образует Ca²⁺-высвобождающие каналы и локализуется в эндоплазматическом ретикулуме. Доминантно-негативные мутации влияют на формирование и поддержание сфинктера радужки, приводя к специфической иридогипоплазии вокруг зрачка и фиксированному мидриазу.
В обзоре литературы по синдрому Жиляспи Ciaccio и соавт. (2024) проанализировали 33 молекулярно подтвержденных случая и подтвердили, что моторное развитие задерживается, но со временем улучшается, умственная отсталость наблюдается не у всех (17% имеют нормальный интеллект), а неврологические признаки являются непрогрессирующими3).
Благодаря распространению технологий полного экзомного секвенирования продолжается идентификация новых мутаций PAX6. По состоянию на 2018 год в базе данных Human PAX6 Mutation Database было зарегистрировано 491 мутация, и с тех пор сообщалось еще о примерно 250 новых мутациях1). Также выявляются случаи, когда мутации в некодирующих областях вызывают аниридию, что позволяет надеяться на объяснение случаев, которые ранее не удавалось диагностировать с помощью стандартных методов9).
При хирургии катаракты у пациентов с тяжелой ААК полезна техника визуализации с помощью хрусталикового ретроиллюминатора4). Этот метод позволяет безопасно выполнять факоэмульсификацию даже у пациентов с ААК 3–4 степени, что приводит к улучшению послеоперационной остроты зрения.
Становится очевидным, что характер прогрессирования ААК различается в зависимости от типа мутации PAX6. Снижение стоимости генетического тестирования делает прогнозирование клинического течения на основе типа мутации и раннее вмешательство реальными вариантами.
В случаях сочетания аниридии и трисомии 21, несмотря на сосуществование обоих заболеваний, сообщалось о относительно легком течении2). Понимание влияния на фенотип при наличии у одного пациента нескольких генетических нарушений может дать важные сведения для будущей персонализированной медицины.
На уровне фундаментальных исследований изучается применение препарата, вызывающего пропуск стоп-кодонов (аталурен), при мутациях типа PTC для лечения аниридии8). Что касается генной терапии PAX6, ведутся фундаментальные исследования по генному замещению с использованием вектора AAV-PAX6 на модели мышей с мутацией Sey. Ожидается переход к клиническим испытаниям в будущем.
Клинические испытания трансплантации листов эпителиальных клеток роговицы, полученных из iPS-клеток, проводятся в Японии и за рубежом, что привлекает внимание как новый метод лечения ААК8). Что касается искусственной радужки (например, CustomFlex Artificial Iris), накоплен опыт использования за рубежом. Контактные линзы с искусственной радужкой как вспомогательное средство подлежат страховому покрытию.
Важной задачей на будущее является накопление данных крупных регистров в Японии для понимания реальной ситуации и повышения качества доказательств8). Ожидается оптимизация прогнозирования прогрессирования ААК на основе индивидуальных генетических мутаций и раннего вмешательства.