La aniridia es una enfermedad congénita rara caracterizada por diversos grados de hipoplasia o ausencia del iris. El término “aniridia” es un nombre inapropiado, ya que con gonioscopia o ultrasonido biomicroscópico (UBM) casi siempre se observan fragmentos de tejido iridiano.
La prevalencia se estima en aproximadamente 1/40,000 a 1/100,000, sin diferencias raciales o de género significativas reportadas 1). En la CIE-10 se clasifica como Q13.1.
Es una enfermedad panocular que afecta no solo al iris, sino también a la córnea, cristalino, ángulo, fóvea y nervio óptico1), presentando diversas complicaciones oculares que amenazan la visión. El pronóstico visual es generalmente desfavorable, con una agudeza visual corregida que a menudo se limita a alrededor de 0,1. El reflejo pupilar está ausente, pero la acomodación se conserva, y entre el 60 y 90% de los casos son bilaterales.
Se reconocen los siguientes tres fenotipos.
Aniridia aislada
Frecuencia: aproximadamente 2/3 del total.
Herencia: autosómica dominante (AD).
Características: causada por mutaciones en el gen PAX6. No se asocia con síntomas sistémicos. La penetrancia es completa, pero la expresividad es variable.
Síndrome WAGR
Frecuencia: parte de los casos esporádicos.
Herencia: deleción contigua de PAX6 y WT1.
Características: asociado a tumor de Wilms, anomalías genitourinarias y retraso mental. El riesgo de tumor es de hasta el 50%.
Síndrome de Gillespie
Frecuencia: aproximadamente el 2% del total.
Herencia: mutación en el gen ITPR1.
Características: se asocia con ataxia cerebelosa y discapacidad intelectual. Es característica una anomalía iridiana específica que presenta midriasis fija3).
La aniridia esporádica representa aproximadamente 1/3 del total y es causada por una deleción de novo en 11p13 que incluye PAX6. Si la deleción se extiende al gen WT1 adyacente, causa el síndrome WAGR1). Entre el 25 y 30% de los casos de aniridia esporádica desarrollan tumor de Wilms, con un riesgo relativo reportado de 67.
PAX6 es el gen maestro del desarrollo ocular y participa en el desarrollo del ojo, tubo neural, bulbo olfatorio, islotes de Langerhans del páncreas y epitelio olfatorio. La enfermedad se produce por pérdida de función de un alelo (haploinsuficiencia), y la anomalía en ambos alelos es letal embrionaria. En 2017 fue designada como enfermedad intratable según la Ley de Enfermedades Intratables, y los casos de gravedad grado III o superior (ver sección de diagnóstico y pruebas) son elegibles para subsidios médicos7).
Q¿Se puede desarrollar aniridia aunque no haya antecedentes familiares?
A
Los casos esporádicos (mutaciones de novo) representan aproximadamente un tercio del total y pueden ocurrir sin antecedentes familiares. En los casos esporádicos existe la posibilidad de síndrome de WAGR, por lo que son importantes las pruebas genéticas y el cribado de tumor de Wilms mediante ecografía abdominal.
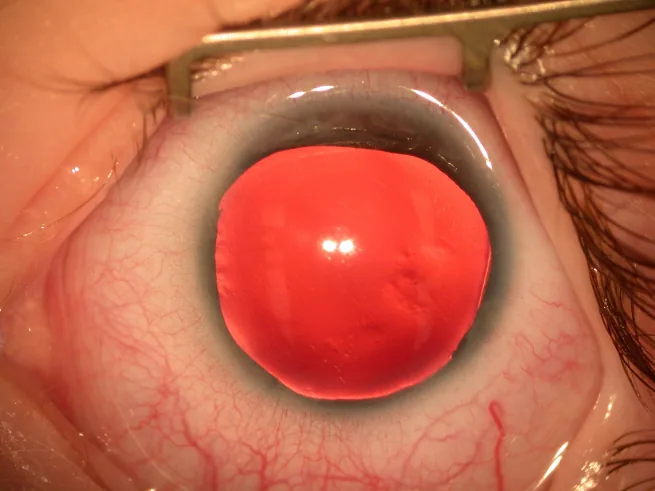
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
En la fotografía del segmento anterior, el iris está casi ausente, con un fino remanente de iris en la periferia corneal. El área pupilar está marcadamente dilatada y se observa un reflejo rojo, lo que representa un hallazgo clínico típico de la aniridia.
El fenotipo varía entre familias y dentro de una misma familia, pero la diferencia entre ambos ojos suele ser pequeña.
Iris: Varía desde una ausencia casi completa hasta una hipoplasia leve. En casos leves, la pupila puede parecer normal, pero se confirma la anomalía mediante transiluminación del iris.
Hipoplasia de la fóvea: Se observa en casi todos los casos y se caracteriza por un reflejo foveal disminuido, hipopigmentación macular y vasos retinianos que cruzan el área foveal. La OCT muestra ausencia de la depresión foveal1).
Cataratas: La opacidad polar anterior congénita es común, y las opacidades que afectan la función visual ocurren finalmente en el 50-85% de los casos. Por lo general, aparecen dentro de los primeros 20 años de vida6). Se estima que se presentan en aproximadamente el 80% de los casos.
Glaucoma: la incidencia reportada varía entre 50 y 75%. Generalmente se presenta en la segunda infancia o en la edad adulta, por lo que es esencial realizar evaluaciones periódicas de la presión intraocular durante la infancia. Se consideran dos mecanismos patogénicos: la alta resistencia al flujo de humor acuoso en la malla trabecular, y la adhesión del iris residual en la periferia extrema a la malla trabecular, que produce un glaucoma de ángulo cerrado.
Queratopatía asociada a aniridia (AAK): causa opacidad corneal progresiva, con una incidencia del 20 al 80%. Se produce engrosamiento corneal periférico y neovascularización que progresa hacia el centro 2). La córnea es más gruesa que en individuos sanos. En la infancia, la córnea es transparente, pero con la edad se produce una invasión conjuntival gradual y se forma un pannus vascularizado.
Subluxación del cristalino: poco frecuente, generalmente se desplaza hacia arriba 1). Las fibras de la zónula de Zinn son frágiles.
La principal causa es la displasia foveal, y la agudeza visual corregida suele ser de 0,1 a 0,2. El pronóstico visual es especialmente malo cuando se asocia a hipoplasia macular. La corrección refractiva y el cuidado de baja visión desde la infancia son importantes para el desarrollo visual.
Dado que PAX6 se expresa no solo en el tejido ocular sino también en el sistema nervioso central, los islotes de Langerhans del páncreas y el epitelio olfatorio, pueden presentarse las siguientes complicaciones extraoculares8).
Tumor de Wilms: en el síndrome WAGR, la tasa de asociación es del 0 al 26.9%; aproximadamente el 30% de los casos esporádicos se presentan antes de los 5 años8)
Anomalías urogenitales: 0 a 33.3%8)
Retraso del desarrollo psicomotor: 0 a 50%8)
Anomalías del sistema nervioso central: agenesia pineal 30%, trastornos graves de la formación cerebral 10%
Agenesia del cuerpo calloso, epilepsia, anosmia, intolerancia a la glucosa: reflejan la expresión multiorgánica de PAX6
Complicaciones sistémicas del síndrome de WAGR: anomalías dentales 35%, anomalías musculoesqueléticas 13%, hiposmia 5%, diabetes 7%, etc.8)
Los factores importantes que determinan la función visual son el glaucoma, la hipoplasia macular, el nistagmo, la queratopatía, las cataratas y la anomalía del iris. Dado que el daño del campo visual y la agudeza visual causado por el glaucoma es irreversible, el control de la presión intraocular es de suma importancia en el seguimiento.8)
La mayoría de los casos de aniridia congénita son causados por mutaciones heterocigotas en el gen PAX6, ubicado en el brazo corto del cromosoma 11 (11p13). La haploinsuficiencia de PAX6 es el principal mecanismo patogénico.1)
El gen PAX6 es un gen maestro del desarrollo ocular y desempeña un papel crucial en el desarrollo del ojo, el tubo neural, el bulbo olfatorio y el páncreas. Se requieren dos copias de PAX6 para el desarrollo ocular normal, y la pérdida de función de una sola copia es suficiente para causar aniridia.1)
En un estudio de cohorte de pacientes chinos, se identificaron mutaciones causales en el gen PAX6 en el 96.9% de los casos 1). En la aniridia típica, se detectan mutaciones que inducen la degradación del ARNm dependiente de codones de terminación prematura (NMD) o deleciones a gran escala en el 96% de los casos 1).
Patológicamente, el músculo liso está ausente excepto en la raíz del iris, y se observa un desarrollo deficiente del ángulo. Hay una disfunción de las células madre epiteliales corneales, que conduce a anomalías en el epitelio y la membrana de Bowman, y se forma un pannus vascularizado.
A continuación se muestra el desglose de las mutaciones de PAX6 que causan el fenotipo de aniridia.
Tipo de mutación
Frecuencia
Mutación sin sentido
Aproximadamente 39%
Mutación de cambio de marco de lectura
Aproximadamente 25%
Mutación de empalme
Aproximadamente 13%
Mutación de sentido erróneo
Aproximadamente 12%
Las mutaciones de tipo read-through (mutaciones de extensión del extremo C) representan aproximadamente el 5% y producen una proteína PAX6 anormalmente alargada debido a la conversión del codón de parada en un codón de traducción 6). Las mutaciones de extensión del extremo C suelen asociarse con hipoplasia grave del iris y deterioro visual severo 1)6).
Las mutaciones genéticas son predominantemente de tipo PTC, aunque también se han reportado mutaciones de sentido erróneo 7). En cuanto a la utilidad de las pruebas genéticas, mediante secuenciación Sanger o NGS se detectan mutaciones en casi el 85% de los casos de aniridia aislada. Además, mediante MLPA o CMA se detectan deleciones dentro del gen PAX6 o en regiones cis-reguladoras en casi el 15% de los casos 8).
Wang (2023) identificó una nueva mutación de cambio de marco c.640_646del (p.R214Pfs*28) y reportó un caso que presentaba ausencia completa del iris, displasia de fóvea, ectopia lentis y desprendimiento de retina1).
Ratna et al. (2022) identificaron una mutación de tipo run-on c.1268A>T (p.*423L) en una familia india. Los afectados presentaron aniridia completa, nistagmo, displasia de fóvea, AAK, subluxación superior del cristalino, miopía alta y atrofia óptica, mostrando un fenotipo grave debido a la extensión del extremo C-terminal6).
En la aniridia esporádica, las deleciones grandes que incluyen PAX6 y el gen WT1 causan el síndrome WAGR. El riesgo de tumor de Wilms en presencia de deleción de WT1 es de hasta el 50%1). Si se sospecha síndrome WAGR, las pruebas genéticas confirman las deleciones de PAX6 y WT1, permitiendo la evaluación del riesgo de tumor de Wilms y el seguimiento del retraso del desarrollo8). La evaluación de la región WT1 mediante pruebas genéticas es esencial; se estima que el 30% de los casos esporádicos desarrollan tumor de Wilms antes de los 5 años. Dado que WT1 se encuentra cerca de PAX6, la deleción del brazo corto del cromosoma 11 (deleción 11p13) que afecta a ambos genes asocia aniridia con tumor de Wilms.
El síndrome de Gillespie es causado por una mutación heterocigótica dominante negativa o una mutación bialélica en el gen ITPR13). Hasta la fecha, se han reportado 37 casos con diagnóstico molecular confirmado, y se sabe que el residuo Gly2554 es un punto caliente3).
La clasificación de gravedad para el reconocimiento de enfermedad intratable se define en los siguientes 4 niveles7).
Gravedad
Definición
Grado I
Afectación de un ojo, el otro sano
Grado II
Afectación de ambos ojos, agudeza visual corregida del mejor ojo ≥ 0.3
Grado III
Afectación de ambos ojos, agudeza visual corregida del mejor ojo ≥ 0.1 y < 0.3
Grado IV
Afectación bilateral, agudeza visual corregida del ojo mejor < 0.1
Incluso en grados I a III, si hay estrechamiento del campo visual debido a glaucoma u otras causas (campo visual central residual dentro de 20 grados con el estímulo Goldmann I/4), se pasa al siguiente grado de gravedad. La gravedad de grado III o superior es elegible para asistencia médica financiera 7).
El diagnóstico clínico es fácil si se confirma la ausencia o hipoplasia del iris con una lámpara de hendidura. La evaluación del tejido iridiano residual se realiza mediante gonioscopia o microscopía ultrasónica biomicroscópica. También se verifica la presencia de anomalías del desarrollo del ángulo de la cámara anterior.
Evaluar sistemáticamente las siguientes complicaciones oculares:
Fóvea hipoplásica: Evaluar mediante OCT la presencia o ausencia de depresión foveal. Es típica una imagen con grosor retiniano normal y ausencia de depresión foveal.
Presión intraocular: Medir periódicamente durante la infancia. Existe un amplio rango de edades de inicio, desde glaucoma congénito hasta glaucoma en la edad adulta.
Córnea: Evaluar la presencia y el grado de progresión de AAK. El grosor corneal central suele estar aumentado6).
Cristalino: verificar la presencia de opacidad del polo anterior, opacidad cortical y subluxación.
Nervio óptico: evaluar la presencia de hipoplasia o coloboma.
El objetivo más importante en la evaluación genética de la aniridia es confirmar si la deleción de PAX6 se extiende al gen WT11). La secuenciación del exoma completo y la técnica MLPA evalúan mutaciones y deleciones en las regiones de PAX6 y WT11)2).
Ecografía abdominal: cribado del tumor de Wilms. En casos esporádicos se recomienda seguimiento cada pocos meses.
RMN craneal: evaluar anomalías en bulbo olfatorio, cerebelo y cuerpo calloso.
Evaluación del desarrollo: valorar funciones cognitivas y conductuales.
Q¿Es siempre necesaria la prueba genética?
A
En la aniridia esporádica, la evaluación del riesgo de tumor de Wilms por deleción del gen WT1 está directamente relacionada con el pronóstico vital 1). Incluso en casos familiares, debido a la variabilidad fenotípica, se recomienda el diagnóstico mediante pruebas genéticas y asesoramiento genético.
No existe un tratamiento curativo para la aniridia. El manejo se centra en la atención de baja visión para maximizar la visión residual y el tratamiento individualizado de cada complicación 8).
El trasplante de córnea para la opacidad del estroma corneal debe considerarse con precaución8).
El trasplante de córnea puede mejorar la visión a corto plazo, pero la mejora es limitada debido a comorbilidades como la hipoplasia macular. A largo plazo, el pronóstico visual es malo debido a la progresión del glaucoma y la disfunción del injerto.
Tasa de rechazo del trasplante de córnea de espesor total: 64% (informe de Kremer)
Tasa de fracaso del injerto en trasplante de espesor total en dos etapas después del trasplante de limbo: 30% (informe de Holland) 12)
Tasa de mantenimiento anatómico del Boston KPro: 77-100% (a medio plazo), 87% (a largo plazo, 4.5 años)
Glaucoma postoperatorio con Boston KPro: alta tasa del 14.3-88%
Formación de membrana retroprotésica después de Boston KPro: 13.3-61%
En la insuficiencia de células madre epiteliales corneales, se considera el tratamiento quirúrgico8).
Trasplante de limbo alogénico: en 12 ojos de 6 casos, la agudeza visual media logMAR mejoró de 1,4 a 0,35 (seguimiento de 64,4 meses)12)
Trasplante de láminas de células epiteliales orales cultivadas: la córnea se aclaró en el 76,4% de 17 ojos y la agudeza visual mejoró en el 88,2%11)
Cuando se complica con opacidad del estroma corneal, la combinación con trasplante de córnea es útil para mejorar la agudeza visual
Se debe tener precaución con los efectos secundarios sistémicos (nefrotoxicidad, alteración de la tolerancia a la glucosa) asociados al uso prolongado de inmunosupresores
La cirugía de cataratas se considera según el grado de opacidad y fotofobia8).
La catarata se desarrolla en un 50-85% de los casos antes de los 20 años. La cirugía se planifica según la intensidad de la opacidad y la fotofobia. Se ha reportado que la agudeza visual mejora en un 66-100% de los casos operados, pero se deben tener en cuenta los siguientes puntos:
Empeoramiento del glaucoma postoperatorio: 26.9-50%
Casos que requieren cirugía de glaucoma: 4-40%
Síndrome de fibrosis anterior (AFS): ocurrió en 6.4% de 155 ojos (todas mujeres) 15)
Riesgo de disminución de la densidad de células endoteliales corneales
La capsulotomía anterior de menos de 6 mm puede funcionar como pupila debido a la fibrosis
Debido a la fragilidad de las zónulas de Zinn, la inserción del lente intraocular requiere una indicación cuidadosa.
Hu et al. (2024) realizaron facoemulsificación asistida por iluminación retrógrada tipo candelabro en 2 casos de aniridia congénita con AAK grave. Aunque la visualización intraoperatoria habitual era difícil debido a la opacidad corneal, la iluminación posterior permitió una visualización clara del cristalino y la cápsula anterior, y la agudeza visual corregida mejoró a 20/200 y 20/1000 respectivamente a las 3 semanas postoperatorias 4).
El glaucoma afecta directamente el pronóstico visual, por lo que debe tratarse de forma activa8).
Tras el inicio del glaucoma, se maneja según el siguiente algoritmo de 5 pasos.
Tratamiento farmacológico: los betabloqueantes, los simpaticomiméticos y los análogos de prostaglandinas (PG) son efectivos. La brimonidina (agonista alfa-adrenérgico) está contraindicada en menores de 2 años debido al riesgo de depresión del sistema nervioso central. Si existe preocupación por daño epitelial corneal, se deben usar formulaciones sin conservantes.
Cirugía de reconstrucción de la vía de drenaje (goniotomía/trabeculotomía): se recomienda como cirugía inicial16). También se ha reportado goniotomía profiláctica. Sin embargo, puede ser ineficaz en casos donde el iris residual cubre la malla trabecular.
Cirugía filtrante (trabeculectomía): solo hay reportes de casos a corto y mediano plazo. Tiende a tener malos resultados en ojos pediátricos, con una tasa de fístula postoperatoria de aproximadamente el 25%13). También se ha reportado glaucoma maligno postoperatorio.
Cirugía de implante de glaucoma (cirugía de derivación con tubo): se pueden usar dispositivos tipo Baerveldt y Ahmed. En ojos fáquicos, se recomienda insertar el tubo en dirección tangencial, no hacia el centro de la córnea. Se espera un buen control de la presión intraocular.
Ciclocoagulación: último recurso. Se ha reportado que la criocoagulación del cuerpo ciliar conduce a fístula en la mayoría de los casos. Debido a la hipoplasia del cuerpo ciliar, el riesgo de fístula es mayor que en ojos sanos.
Q¿El tratamiento del glaucoma en la aniridia es diferente al del glaucoma habitual?
A
Debido a la anomalía del desarrollo del ángulo, se requiere un enfoque diferente al del glaucoma de ángulo abierto habitual. La primera opción es la cirugía reconstructiva del tracto de salida, seguida de la cirugía de derivación con tubo como una buena alternativa. La brimonidina está contraindicada en menores de 2 años, y el uso de antimetabolitos puede empeorar la AAK, por lo que se requiere una evaluación cuidadosa 8).
La atención de baja visión debe iniciarse de forma temprana8).
La corrección refractiva es fundamental; la tasa de miopía asociada es superior al 64%.
Ayudas ópticas para la visión: gafas correctoras, lupas, gafas de sol, gafas para baja visión (tipo montura y enfoque ajustable)
Ayudas no ópticas para la visión: libros de texto ampliados, atriles, ajuste de iluminación, tiposcopio, tabletas, magnificadores de lectura17)
Durante la edad escolar, es necesaria la selección de ayudas visuales según la función visual, el entrenamiento en su uso y la adecuación del entorno de aprendizaje
Es importante la coordinación con instituciones de bienestar, educación y empleo según la etapa de la vida
El tratamiento de la fotofobia es importante para preservar el desarrollo visual y la calidad de vida8).
Gafas de protección solar: seleccionar y prescribir el color que más reduzca la fotofobia mediante lentes de prueba.
Lentes de contacto blandas (SCL) con iris artificial: tienen un efecto reductor del nistagmo. El tratamiento temprano de protección contra la luz puede contribuir a mejorar la fijación y el desarrollo visual.
Usar sombreros de ala ancha o sombrillas al aire libre, e iluminación indirecta en interiores.
Implante de iris artificial: también existe la opción de insertar un iris artificial durante la cirugía de cataratas5).
Cuando se usan lentes de contacto, se debe realizar un seguimiento cuidadoso de la presencia de daño en las células madre del epitelio corneal.
La mayoría de los pacientes pueden asistir a escuelas regulares, pero necesitan apoyo como libros de texto ampliados, tabletas y atriles. También es una opción asistir a clases para niños con baja visión o utilizar servicios de asesoramiento sobre crianza y educación ofrecidos por escuelas para ciegos o escuelas de apoyo visual especial.
Desde abril de 2017, esta enfermedad ha sido designada como enfermedad rara específica, por lo que incluso si no se tiene un certificado de discapacidad, los pacientes con gravedad de grado III o superior son elegibles para asistencia médica y suministro de ayudas ortopédicas 7). Las ayudas ortopédicas cubiertas incluyen gafas correctivas, gafas de sol, lentes de contacto (incluyendo las de iris artificial), gafas para baja visión, bastones de seguridad para personas con discapacidad visual y ojos protésicos.
6. Fisiopatología y mecanismo detallado de aparición
PAX6 abarca 22 kb de ADN genómico que incluye 14 exones y codifica 422 aminoácidos 1). Tiene dos dominios de unión al ADN (dominio emparejado y homeodominio emparejado), y el dominio PST (rico en prolina, serina y treonina) en el extremo C funciona como activador transcripcional.
PAX6 regula la proliferación, diferenciación, migración y adhesión celular, y sus dianas incluyen el propio PAX6, así como genes que codifican cristalinas del cristalino y queratinas corneales. La expresión continúa en la retina, el cristalino y la córnea del adulto. El gen PAX6 es uno de los genes maestros que controlan la diferenciación de órganos durante el período embrionario.
La mayoría de las mutaciones de PAX6 causan haploinsuficiencia a través de la degradación de ARNm dependiente de mutaciones sin sentido (NMD) 1). Las mutaciones que introducen un codón de terminación prematuro (PTC) (mutaciones sin sentido, de cambio de marco y la mayoría de las mutaciones de empalme) producen el fenotipo típico de aniridia.
Por otro lado, si el PTC se localiza en el exón final o dentro de los últimos 50 pb del penúltimo exón, puede escapar de NMD y traducirse una proteína truncada, lo que podría dar lugar a un fenotipo grave1).
Se ha reportado un caso raro de un paciente con la mutación sin sentido c.282C>A (p.Cys94*) en PAX6 y trisomía 21. La mutación PAX6 ocurrió de novo y el paciente presentó aniridia bilateral completa, glaucoma congénito, AAK y displasia de fóvea2).
En la serie de gonioscopia de Grant y Walton, se demostró que el estroma del iris se extiende anteriormente sobre el trabeculado formando adherencias tempranas, que progresan a una capa y finalmente a la oclusión del ángulo14). Este mecanismo es el principal factor en el desarrollo del glaucoma. Patológicamente, la base es un defecto del músculo liso que deja la raíz del iris y un desarrollo deficiente del ángulo.
La AAK es causada principalmente por deficiencia de células madre limbares (LSCD), pero también están implicadas la diferenciación anormal del epitelio corneal, la adhesión anormal, la infiltración de células conjuntivales y la producción insuficiente de lágrimas. La deficiencia de metaloproteinasa de matriz 9 (MMP-9), regulada por PAX6, provoca acumulación de fibrina e infiltración de células inflamatorias, y la pérdida de transparencia se debe a la alteración de la disposición del colágeno en el estroma.
La AAK se clasifica en 5 etapas. La etapa I muestra solo anomalías del epitelio periférico, la etapa II cambios epiteliales centrípetos (sin alcanzar el centro), la etapa III cambios epiteliales en la córnea central y neovascularización superficial periférica, la etapa IV neovascularización superficial de toda la córnea, y la etapa V anomalías epiteliales en toda la córnea y cicatrización estromal profunda 10).
Existe una relación entre el estado de la mutación de PAX6 y la progresión de la AAK. En pacientes con mutaciones PTC o de extensión del extremo C, la AAK progresa de forma dependiente de la edad, mientras que otros tipos de mutaciones pueden presentar queratopatía no progresiva 11).
El síndrome de Gillespie es causado por mutaciones en el gen ITPR1 3). ITPR1 es miembro de la familia de receptores de IP3, forma canales de liberación de Ca²⁺ y se localiza en el retículo endoplásmico. Las mutaciones dominantes negativas afectan la formación y el mantenimiento del músculo esfínter del iris, provocando una displasia específica del iris alrededor de la pupila y midriasis fija.
En la revisión bibliográfica del síndrome de Gillespie de Ciaccio et al. (2024), basada en el análisis de 33 casos confirmados molecularmente, se confirmó que el desarrollo motor se retrasa pero mejora con el tiempo, que la discapacidad intelectual no está presente en todos los casos (17% tienen inteligencia normal) y que los signos neurológicos no son progresivos 3).
Con la difusión de la tecnología de secuenciación del exoma completo, se continúan identificando nuevas mutaciones en PAX6. En 2018, la Base de Datos de Mutaciones de PAX6 Humano registraba 491 mutaciones, y desde entonces se han reportado aproximadamente 250 nuevas mutaciones 1). También se están identificando casos de aniridia causados por mutaciones en regiones no codificantes, lo que se espera que aclare casos que no pudieron diagnosticarse con pruebas convencionales 9).
En casos de cirugía de cataratas con AAK grave, la técnica de visualización asistida por iluminación invertida tipo candelabro es útil 4). Este procedimiento permite realizar una facoemulsificación segura incluso en pacientes con AAK de grado 3 a 4, logrando una mejora en la agudeza visual postoperatoria.
Se está aclarando que el patrón de progresión de la AAK difiere según el tipo de mutación de PAX6. Con la reducción de costos de las pruebas genéticas, la predicción del curso clínico basada en el tipo de mutación y la intervención temprana se están convirtiendo en opciones realistas.
En casos de aniridia combinada con trisomía 21, se han reportado ejemplos con un curso relativamente leve a pesar de la coexistencia de ambas enfermedades2). Comprender el impacto en el fenotipo cuando múltiples trastornos genéticos coexisten en un mismo paciente podría proporcionar conocimientos importantes para la medicina personalizada futura.
Se está evaluando a nivel de investigación básica la aplicación de ataluren, un fármaco de lectura forzada para mutaciones de tipo PTC, en la aniridia8). En cuanto a la terapia génica de PAX6, se están realizando investigaciones básicas de suplementación génica con vectores AAV-PAX6 en modelos de ratón con mutación Sey. Se espera su desarrollo hacia ensayos clínicos futuros.
Se están realizando ensayos clínicos de trasplante de láminas de células epiteliales corneales derivadas de células iPS tanto en Japón como en el extranjero, y se espera que sea un nuevo tratamiento para la AAK 8). En cuanto al iris artificial (como el CustomFlex Artificial Iris), se han acumulado experiencias de uso en el extranjero. Como dispositivo de ayuda, las lentes de contacto con iris artificial están cubiertas por el seguro médico.
La acumulación de datos de registros a gran escala en Japón para comprender la situación real y mejorar la calidad de la evidencia son temas importantes para el futuro 8). Se espera poder predecir la progresión de la AAK basándose en mutaciones genéticas individuales y optimizar la intervención temprana.
Wang Q, Wei WB, Shi XY, Rong WN.. A novel PAX6 variant as the cause of aniridia in a Chinese patient with SRRRD. BMC Med Genomics. 2023;16(1):182. doi:10.1186/s12920-023-01620-w. PMID:37542296; PMCID:PMC10401864.
Vasilyeva TA, Sukhanova NV, Marakhonov AV, Kuzina NY, Shilova NV, Kadyshev VV, et al. Co-Occurrence of Congenital Aniridia Due to Nonsense PAX6 Variant p.(Cys94*) and Chromosome 21 Trisomy in the Same Patient. International journal of molecular sciences. 2023;24(21). doi:10.3390/ijms242115527. PMID:37958513; PMCID:PMC10650867.
Ciaccio C, Taddei M, Pantaleoni C, Grisoli M, Di Bella D, Magri S, Taroni F, D’Arrigo S. Phenotypic Spectrum and Natural History of Gillespie Syndrome. An Updated Literature Review with 2 New Cases. Cerebellum (London, England). 2024;23(6):2655-2670. doi:10.1007/s12311-024-01733-7. PMID:39177731; PMCID:PMC11585489.
Hu J, Hu CC. Chandelier retroillumination-assisted cataract surgery in two cases of congenital aniridia with severe aniridia-associated keratopathy: case series. Therapeutic advances in ophthalmology. 2024;16:25158414241302879. doi:10.1177/25158414241302879. PMID:39619855; PMCID:PMC11605765.
Christodoulou E, Batsos G, Parikakis E, Papadopoulos V, Karagiannis D, Koumoutsos PP, et al. Combined Post-Traumatic Total Aniridia and Glaucoma Management. Case reports in ophthalmology. 2021;12(1):204-207. doi:10.1159/000511099. PMID:33976683; PMCID:PMC8077530.
Ratna R, Tibrewal S, Gour A, Gupta R, Mathur U, Vanita V. A rare case of congenital aniridia with an unusual run-on mutation in PAX6 gene. Indian journal of ophthalmology. 2022;70(7):2661-2664. doi:10.4103/ijo.IJO_433_22. PMID:35791194; PMCID:PMC9426074.
Lim HT, Kim DH, Kim H. PAX6 aniridia syndrome: clinics, genetics, and therapeutics. Current opinion in ophthalmology. 2017;28(5):436-447. doi:10.1097/ICU.0000000000000405. PMID:28598868.
Edén U, Riise R, Tornqvist K.. Corneal involvement in congenital aniridia. Cornea. 2010;29(10):1096-1102. doi:10.1097/ico.0b013e3181d20493. PMID:20567200.
Nishida K, Kinoshita S, Ohashi Y, Kuwayama Y, Yamamoto S.. Ocular surface abnormalities in aniridia. Am J Ophthalmol. 1995;120(3):368-375. doi:10.1016/s0002-9394(14)72167-1. PMID:7661209.
Holland EJ, Djalilian AR, Schwartz GS. Management of aniridic keratopathy with keratolimbal allograft: a limbal stem cell transplantation technique. Ophthalmology. 2003;110(1):125-30. doi:10.1016/s0161-6420(02)01451-3. PMID:12511357.
Wiggins RE, Tomey KF.. The results of glaucoma surgery in aniridia. Arch Ophthalmol. 1992;110(4):503-505. doi:10.1001/archopht.1992.01080160081036. PMID:1562257.
Grant WM, Walton DS. Progressive changes in the angle in congenital aniridia, with development of glaucoma. American journal of ophthalmology. 1974;78(5):842-7. doi:10.1016/0002-9394(74)90308-0. PMID:4423758.
Tsai JH, Freeman JM, Chan CC, Schwartz GS, Derby EA, Petersen MR, et al. A progressive anterior fibrosis syndrome in patients with postsurgical congenital aniridia. American journal of ophthalmology. 2005;140(6):1075-9. doi:10.1016/j.ajo.2005.07.035. PMID:16376654.
Adachi M, Dickens CJ, Hetherington J Jr, Hoskins HD, Iwach AG, Wong PC, et al. Clinical experience of trabeculotomy for the surgical treatment of aniridic glaucoma. Ophthalmology. 1997;104(12):2121-5. doi:10.1016/s0161-6420(97)30041-4. PMID:9400774.
Vincent SJ. The use of contact lenses in low vision rehabilitation: optical and therapeutic applications. Clinical & experimental optometry. 2017;100(5):513-521. doi:10.1111/cxo.12562. PMID:28664572.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.