Hormona de crecimiento
Frecuencia: Necesaria en aproximadamente el 70% de los pacientes con ONH.
Indicación: Iniciar cuando se confirme la deficiencia de hormona de crecimiento.

La hipoplasia del nervio óptico (ONH) es la anomalía congénita del nervio óptico más frecuente, caracterizada por una reducción en el número de axones del nervio óptico. Puede ocurrir unilateral o bilateralmente y puede asociarse con defectos estructurales de la línea media cerebral.
Briere la describió histológicamente por primera vez en 1877, y Reeves proporcionó una descripción clínica en 1941. En 1956, de Morsier informó su asociación con la ausencia del septum pellucidum, dando lugar al término síndrome de de Morsier (displasia septo-óptica, SOD). En 1970, Hoyt et al. informaron características clínicas detalladas, aumentando el conocimiento de esta afección.
La SOD se diagnostica cuando están presentes dos o más de los siguientes tres hallazgos3)6):
La prevalencia de SOD se estima en aproximadamente 1 de cada 10,000 nacimientos2)6). Epidemiológicamente, es la tercera causa más común de discapacidad visual en niños menores de 3 años. Se han reportado tasas de 10.9 por 100,000 habitantes en Inglaterra y 17.3 en Suecia.
En un estudio de 16 casos en la Universidad de Niigata en Japón, la mediana de edad en la primera visita fue de 2.4 años, 12/16 (75%) eran mujeres y 11/16 (69%) eran bilaterales 1).
Una forma grave es la aplasia del nervio óptico (optic nerve aplasia). La papila y los vasos retinianos están completamente ausentes, y no hay percepción de luz.
La hipoplasia segmentaria superior del nervio óptico (superior segmental optic hypoplasia; SSOH) es un tipo especial en el que solo las fibras del nervio óptico superiores son hipoplásicas, y se ha señalado una asociación con diabetes materna. La prevalencia en Japón se reporta en aproximadamente 0.3%. No hay diferencia de sexo.
ONH se refiere a una anomalía morfológica del nervio óptico solo. SOD es un síndrome que cumple dos o más de la tríada de ONH, disfunción hipofisaria y anomalías estructurales del cerebro medio; ONH es uno de los componentes de SOD. Se ha reportado que aproximadamente el 37.5% de los pacientes con ONH cumplen los criterios diagnósticos de SOD 1).
La agudeza visual en ONH varía desde normal hasta ausencia de percepción de luz. Muchos casos tienen agudeza de 0.1 o menos, y la agudeza visual depende de la densidad del haz papilomacular. La hipoplasia del nervio óptico difiere de otras anomalías congénitas de la papila en este aspecto: incluso si la mácula está formada, el grado de desarrollo del haz de fibras nerviosas papilomaculares varía, por lo que la agudeza visual varía desde 1.0 hasta extremadamente baja.
En datos japoneses, la mala visión se encontró en 11/16 casos (69%), estrabismo en 8/16 casos (50%) y nistagmo en 5/16 casos (31%)1).
Se observan hallazgos característicos en la oftalmoscopia.
Las frecuencias de los principales hallazgos oculares y complicaciones sistémicas se muestran a continuación.
| Hallazgo | Frecuencia |
|---|---|
| Anomalías estructurales del SNC | Aproximadamente 90% |
| Trastornos del neurodesarrollo | Aproximadamente 70% |
| Disfunción hipotalámica (bilateral) | 81% |
| Disfunción hipotalámica (unilateral) | 69% |
| Retraso del desarrollo (bilateral) | 78% |
Incluso en casos unilaterales, se encuentra disfunción hipotalámica en el 69%, y existen anomalías cerebrales en el 18.2% de los pacientes asintomáticos1). La resonancia magnética y el cribado endocrino son esenciales incluso en casos unilaterales.
La patología de la ONH es un fallo en el desarrollo de las células ganglionares de la retina (CGR) y las fibras nerviosas. Existen dos hipótesis: la teoría de la anomalía del desarrollo y la teoría de la degeneración retrógrada. También se ha propuesto la hipótesis de que la isquemia del quiasma óptico y del nervio óptico debida a trastornos vasculares de la arteria cerebral anterior es la causa.
La mayoría de los casos son esporádicos2)3).
Las siguientes mutaciones genéticas pueden estar implicadas.
El diagnóstico de ONH se basa en los hallazgos oftalmoscópicos, combinados con diagnóstico por imagen y cribado endocrino.
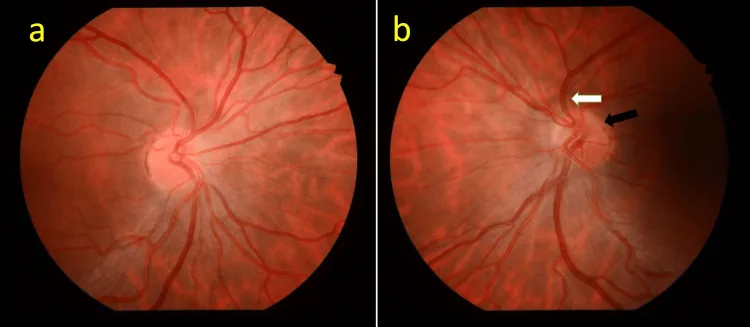
La confirmación del signo del doble anillo es el primer paso del diagnóstico. Una relación DM/DD de 3 o más (3.2 o más indica papila pequeña) sirve como referencia.
En un estudio de pacientes japoneses, se encontraron anomalías cerebrales en el 43.8% y SOD en el 37.5%. Es de destacar que, incluso en pacientes asintomáticos, el 18.2% presentaba anomalías cerebrales 1). Además, en 2 de 3 pacientes con disfunción hipofisaria, la resonancia magnética mostró una morfología hipofisaria normal 1).
Se recomiendan las siguientes pruebas para todos los pacientes con ONH:
Es necesario el diagnóstico diferencial con las siguientes enfermedades:
Sí. En un estudio de pacientes japoneses, 2 de cada 3 pacientes con hipopituitarismo tenían hallazgos normales en la RM1). Se debe realizar un cribado endocrino en todos los pacientes independientemente de los resultados de la RM.
No existe un tratamiento curativo para la ONH en sí. El manejo se centra en optimizar la función visual y abordar las complicaciones sistémicas (especialmente las anomalías endocrinas). Es esencial la colaboración de un equipo multidisciplinario (oftalmología, endocrinología, pediatría, neurología, rehabilitación). Se recomienda evaluar el crecimiento cada seis meses y la función visual anualmente.
Hormona de crecimiento
Frecuencia: Necesaria en aproximadamente el 70% de los pacientes con ONH.
Indicación: Iniciar cuando se confirme la deficiencia de hormona de crecimiento.
Hormona tiroidea
Frecuencia: Necesaria en aproximadamente el 43%.
Indicación: Iniciar el reemplazo cuando TSH/FT4 sean anormales.
Corticosteroides suprarrenales
Frecuencia: Requerida en aproximadamente el 27% de los casos.
Precaución: La insuficiencia suprarrenal puede ser mortal durante el estrés. Es esencial instruir sobre la dosificación de estrés (aumento de la dosis durante fiebre o cirugía)5).
Hormona antidiurética
Frecuencia: Aproximadamente el 5% presenta diabetes insípida.
Precaución: La corrección rápida del sodio puede provocar convulsiones. La velocidad de corrección debe ser inferior a 0.5 mEq/L/hora4).
En casos de manejo en adultos, se han reportado regímenes de reemplazo como levotiroxina 137 μg, desmopresina e hidrocortisona 10 mg (mañana)/7.5 mg (tarde)2).
La hipoplasia del nervio óptico en sí misma no es progresiva a menos que se desarrolle glaucoma. En casos sin glaucoma, se deben evitar gotas oftálmicas o tratamientos quirúrgicos innecesarios para reducir la presión intraocular. Sin embargo, las anomalías endocrinas pueden aparecer o empeorar con el tiempo, por lo que el seguimiento a largo plazo es esencial1). El diagnóstico temprano y el inicio de la terapia de reemplazo hormonal antes de los 3 años en casos necesarios pueden prevenir secuelas, por lo que es importante tener en cuenta esta enfermedad incluso en casos unilaterales y no pasarla por alto.
La hipoplasia del nervio óptico en sí misma no es progresiva y la visión suele ser estable a menos que se desarrolle glaucoma. Sin embargo, las anomalías endocrinas pueden aparecer más tarde, por lo que es importante continuar con evaluaciones sistémicas periódicas.
La esencia de la ONH es una reducción de la capa de fibras nerviosas de la retina (CFNR) y de las células ganglionares, con poco efecto sobre las capas externas de la retina. Hay dos hipótesis principales sobre el mecanismo de desarrollo.
También existe la hipótesis de que la isquemia en la región del quiasma óptico y el nervio óptico debida a trastornos vasculares de la arteria cerebral anterior puede estar involucrada.
La disfunción hipotalámica se observa en el 69% de los casos unilaterales y en el 81% de los bilaterales. La glándula pituitaria y el nervio óptico están próximos embriológicamente, y se cree que el mismo trastorno del desarrollo afecta a ambos.
El retraso del desarrollo se observa en el 75% de todos los casos, con una tasa más alta en los bilaterales (78%) que en los unilaterales (39%).
SOD plus es una condición que incluye malformaciones corticales (p. ej., polimicrogiria, esquizencefalia) además de la SOD clásica, y se ha informado que ocurre con mayor frecuencia que la SOD clásica 7). El pronóstico del neurodesarrollo es peor y el riesgo de epilepsia es mayor.
Está en curso un ensayo clínico (NCT06760546) de setmelanotide, un agonista del receptor de melanocortina 4 (MC4R), como terapia farmacológica para la obesidad asociada a la SOD 2). La obesidad hipotalámica reduce significativamente la calidad de vida de los pacientes con SOD, por lo que se está prestando atención como una nueva opción de tratamiento.
En neonatos con SOD diagnosticados con normoglucemia, se han reportado el manejo del micropene y criptorquidia con terapia de testosterona (25 mg intramuscular, una vez al mes durante 3 meses) y ensayos de terapia con FSH recombinante 5). Estos indican el potencial de la intervención endocrina temprana, pero la eficacia y seguridad a largo plazo aún no están establecidas.