Wachstumshormon
Häufigkeit : Bei etwa 70 % der ONH-Patienten erforderlich.
Indikation : Beginn bei bestätigtem Wachstumshormonmangel.

Die Optikushypoplasie (ONH) ist die häufigste angeborene Anomalie des Sehnervs, gekennzeichnet durch eine verminderte Anzahl von Sehnervenaxonen. Sie kann ein- oder beidseitig auftreten und mit Defekten der Mittellinienstrukturen des Gehirns einhergehen.
Erstmals 1877 von Briere histologisch beschrieben, erfolgte die klinische Beschreibung 1941 durch Reeves. 1956 berichtete de Morsier über die Assoziation mit dem Fehlen des Septum pellucidum, bekannt als de-Morsier-Syndrom (septo-optische Dysplasie, SOD). 1970 veröffentlichten Hoyt et al. detaillierte klinische Beschreibungen, die zur weiteren Anerkennung dieser Erkrankung führten.
Die SOD wird diagnostiziert, wenn mindestens zwei der folgenden drei Kriterien erfüllt sind3)6):
Die Prävalenz der SOD wird auf etwa 1/10.000 Geburten geschätzt2)6). Epidemiologisch ist sie die dritthäufigste Ursache für Sehbehinderung bei Kindern unter 3 Jahren. In England wird eine Rate von 10,9 pro 100.000 Einwohner berichtet, in Schweden von 17,3.
In einer Studie mit 16 Fällen an der Universität Niigata in Japan betrug das mediane Alter bei Erstdiagnose 2,4 Jahre, 12/16 (75 %) waren weiblich und 11/16 (69 %) waren bilateral 1).
Eine schwere Form ist die Optikusaplasie (optic nerve aplasia). Die Papille und die Netzhautgefäße fehlen vollständig, und es besteht keine Lichtwahrnehmung.
Die superiore segmentale Optikushypoplasie (superior segmental optic hypoplasia; SSOH) ist eine Sonderform, bei der nur die oberen Sehnervenfasern hypoplastisch sind, und es wurde ein Zusammenhang mit mütterlichem Diabetes festgestellt. Die Prävalenz in Japan beträgt etwa 0,3 %. Es gibt keine Geschlechterunterschiede.
ONH bezeichnet eine isolierte morphologische Anomalie des Sehnervs. SOD ist ein Syndrom, das mindestens zwei der drei Kriterien erfüllt: ONH, Hypophyseninsuffizienz und Mittellinien-Hirnanomalien. ONH ist eine Komponente der SOD. Einem Bericht zufolge erfüllen etwa 37,5 % der ONH-Patienten die diagnostischen Kriterien für SOD 1).
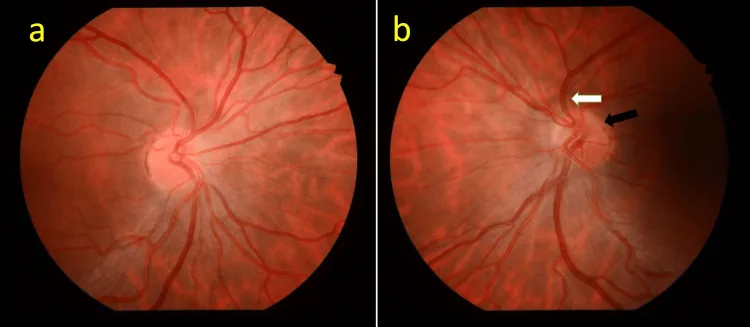
Die Sehschärfe bei ONH reicht von normal bis zu keiner Lichtwahrnehmung. In den meisten Fällen liegt sie unter 0,1 und hängt von der Dichte des papillomakulären Bündels ab. Die Optikushypoplasie unterscheidet sich in diesem Punkt von anderen angeborenen Papillenanomalien: Auch wenn die Makula ausgebildet ist, variiert der Entwicklungsgrad des papillomakulären Nervenfaserbündels, was zu einer breiten Spanne der Sehschärfe von 1,0 bis sehr niedrig führt.
Nach japanischen Daten lag eine Sehschwäche bei 11/16 Fällen (69 %) vor, Strabismus bei 8/16 Fällen (50 %) und Nystagmus bei 5/16 Fällen (31 %)1).
Ophthalmoskopisch zeigen sich charakteristische Befunde.
Die wichtigsten Augenmerkmale und die Häufigkeit systemischer Komplikationen sind unten aufgeführt.
| Befund | Häufigkeit |
|---|---|
| Strukturelle ZNS-Anomalien | Etwa 90 % |
| Neurologische Entwicklungsstörungen | Etwa 70 % |
| Hypothalamische Dysfunktion (beidseitig) | 81% |
| Hypothalamische Dysfunktion (einseitig) | 69% |
| Entwicklungsverzögerung (beidseitig) | 78% |
Auch bei einseitigem Befund liegt in 69% eine hypothalamische Dysfunktion vor, und bei 18,2% der asymptomatischen Patienten bestehen Hirnanomalien1). MRT und endokrinologisches Screening sind auch bei einseitigem Befund obligat.
Die Pathologie der ONH ist eine Dysgenesie retinaler Ganglienzellen (RGC) und Nervenfasern. Es gibt zwei Hypothesen: die Dysgenesie-Hypothese und die retrograde Degenerationshypothese. Auch eine Hypothese, die eine Ischämie des Chiasmas und des Sehnervs durch vaskuläre Störungen der vorderen Hirnarterie als Ursache vorschlägt, wurde aufgestellt.
Die meisten Fälle sind sporadisch2)3).
Folgende Genmutationen können beteiligt sein.
Die Diagnose der ONH basiert auf ophthalmoskopischen Befunden und wird mit Bildgebung und endokrinologischem Screening kombiniert.
Die Bestätigung des Doppelringzeichens ist der erste Schritt der Diagnose. Ein DM/DD-Verhältnis ≥ 3 (≥ 3,2 für kleine Papille) ist ein Richtwert.
In einer Studie mit japanischen Patienten wurden bei 43,8 % Hirnanomalien und bei 37,5 % SOD festgestellt. Bemerkenswert ist, dass bei 18,2 % der asymptomatischen Patienten Hirnanomalien vorlagen 1). Darüber hinaus war bei 2 von 3 Patienten mit Hypophysenunterfunktion die Hypophysenmorphologie im MRT normal 1).
Bei allen ONH-Patienten werden die folgenden Untersuchungen empfohlen.
Eine Abgrenzung zu folgenden Erkrankungen ist erforderlich.
Ja, das ist möglich. In einer Studie mit japanischen Patienten hatten 2 von 3 Patienten mit Hypophyseninsuffizienz normale MRT-Befunde 1). Unabhängig vom MRT-Ergebnis sollte bei allen Patienten ein endokrines Screening durchgeführt werden.
Es gibt keine kurative Behandlung für ONH selbst. Der Schwerpunkt der Behandlung liegt auf der Optimierung der Sehfunktion und der Behandlung systemischer Komplikationen (insbesondere endokriner Anomalien). Eine multidisziplinäre Zusammenarbeit (Augenheilkunde, Endokrinologie, Pädiatrie, Neurologie, Rehabilitation) ist unerlässlich. Eine Beurteilung des Wachstums alle sechs Monate und der Sehfunktion einmal jährlich wird empfohlen.
Wachstumshormon
Häufigkeit : Bei etwa 70 % der ONH-Patienten erforderlich.
Indikation : Beginn bei bestätigtem Wachstumshormonmangel.
Schilddrüsenhormon
Häufigkeit : Bei etwa 43 % erforderlich.
Indikation : Beginn bei TSH- und FT4-Anomalien.
Nebennierenrindenhormon (Kortikosteroid)
Häufigkeit: In etwa 27 % der Fälle erforderlich.
Achtung: Eine Nebenniereninsuffizienz kann in Stresssituationen tödlich sein. Die Anleitung zum Stress-Dosing (Dosiserhöhung bei Fieber/Operationen) ist unerlässlich 5).
Antidiuretisches Hormon
Häufigkeit: Etwa 5 % entwickeln einen Diabetes insipidus.
Achtung: Eine schnelle Korrektur des Natriums kann Krampfanfälle auslösen. Die Korrekturgeschwindigkeit sollte unter 0,5 mEq/L/h liegen 4).
Bei Erwachsenen wurden Substitutionsschemata wie Levothyroxin 137 µg, Desmopressin, Hydrocortison 10 mg (morgens)/7,5 mg (abends) berichtet 2).
Die Optikushypoplasie selbst ist ohne begleitendes Glaukom nicht progredient. Ohne Glaukom sollte auf eine leichtfertige augendrucksenkende Augentropfen- oder Operationstherapie verzichtet werden. Endokrine Anomalien können jedoch im Laufe der Zeit auftreten oder sich verschlechtern, daher ist eine langfristige Nachsorge unerlässlich 1). Eine frühzeitige Diagnose und der Beginn einer Hormonersatztherapie vor dem 3. Lebensjahr in notwendigen Fällen verhindern Folgeschäden, daher ist es wichtig, diese Erkrankung auch bei einseitigem Befall im Hinterkopf zu behalten und nicht zu übersehen.
Die Optikushypoplasie selbst ist nicht progredient, und ohne Glaukom bleibt das Sehvermögen oft stabil. Allerdings können endokrine Anomalien später auftreten, daher ist eine regelmäßige systemische Beurteilung wichtig.
Das Wesen der ONH ist eine Abnahme der retinalen Nervenfaserschicht (RNFL) und der Ganglienzellen, mit geringer Auswirkung auf die äußeren Netzhautschichten. Es gibt zwei Haupthypothesen zum Entstehungsmechanismus.
Es gibt auch die Hypothese, dass eine Ischämie der Sehnervenkreuzungs- und Sehnervenregion aufgrund einer vaskulären Störung der vorderen Hirnarterie beteiligt ist.
Eine hypothalamische Dysfunktion tritt bei 69 % der einseitigen ONH und bei 81 % der beidseitigen ONH auf. Die Hypophyse und der Sehnerv liegen entwicklungsgeschichtlich nahe beieinander, und es wird angenommen, dass dieselbe Entwicklungsstörung beide betrifft.
Eine Entwicklungsverzögerung tritt bei 75 % aller Fälle auf, wobei die Rate bei beidseitigen Fällen (78 %) höher ist als bei einseitigen (39 %).
SOD plus ist ein Zustand, der zusätzlich zur klassischen SOD kortikale Dysplasien (Polymikrogyrie, Schizenzephalie usw.) umfasst und laut einigen Berichten häufiger auftritt als die klassische SOD7). Die neurologische Entwicklungsprognose ist schlechter und das Risiko für Epilepsie ist ebenfalls höher.
Als medikamentöse Therapie für mit SOD verbundene Fettleibigkeit läuft eine klinische Studie (NCT06760546) mit Setmelanotid, einem Melanocortin-4-Rezeptor (MC4R)-Agonisten2). Hypothalamische Fettleibigkeit beeinträchtigt die Lebensqualität von SOD-Patienten erheblich, daher wird sie als neue Behandlungsoption angesehen.
Bei SOD-Neugeborenen mit normalem Blutzucker wurde über die Behandlung von Mikropenis und Kryptorchismus mit Testosterontherapie (25 mg i.m., einmal monatlich für 3 Monate) und Versuche mit rekombinanter FSH-Therapie berichtet 5). Diese zeigen das Potenzial einer frühzeitigen endokrinen Substitution, jedoch sind die langfristige Wirksamkeit und Sicherheit noch nicht etabliert.