Ormone della crescita
Frequenza : Necessario in circa il 70% dei pazienti con ONH.
Indicazione : Iniziare in caso di deficit confermato di ormone della crescita.

L’ipoplasia del nervo ottico (ONH) è l’anomalia congenita del nervo ottico più frequente, caratterizzata da una riduzione del numero di assoni ottici. Può essere unilaterale o bilaterale e accompagnarsi a difetti delle strutture cerebrali mediane.
Descritta istologicamente per la prima volta da Briere nel 1877, la descrizione clinica fu fornita da Reeves nel 1941. Nel 1956 de Morsier riportò l’associazione con l’assenza del setto pellucido, nota come sindrome di de Morsier (displasia setto-ottica, SOD). Nel 1970 Hoyt et al. pubblicarono descrizioni cliniche dettagliate, ampliando il riconoscimento di questa malattia.
La SOD viene diagnosticata quando sono presenti almeno due dei seguenti tre elementi3)6):
La prevalenza della SOD è stimata in circa 1/10.000 nati2)6). Dal punto di vista epidemiologico, è la terza causa di deficit visivo nei bambini di età inferiore a 3 anni. In Inghilterra è stata riportata una prevalenza di 10,9 per 100.000 abitanti, in Svezia di 17,3.
In uno studio di 16 casi presso l’Università di Niigata in Giappone, l’età mediana alla prima diagnosi era di 2,4 anni, 12/16 (75%) erano femmine e 11/16 (69%) erano bilaterali 1).
Una forma grave è l’aplasia del nervo ottico (optic nerve aplasia). La papilla e i vasi retinici sono completamente assenti e non c’è percezione luminosa.
L’ipoplasia segmentale superiore del nervo ottico (superior segmental optic hypoplasia; SSOH) è una forma speciale in cui solo le fibre nervose ottiche superiori sono ipoplasiche, ed è stata segnalata un’associazione con il diabete materno. La prevalenza in Giappone è di circa lo 0,3%. Non ci sono differenze di sesso.
L’ONH si riferisce a un’anomalia morfologica isolata del nervo ottico. La SOD è una sindrome che soddisfa almeno due dei tre criteri: ONH, insufficienza ipofisaria e anomalie cerebrali della linea mediana. L’ONH è una componente della SOD. Secondo un rapporto, circa il 37,5% dei pazienti con ONH soddisfa i criteri diagnostici per la SOD 1).
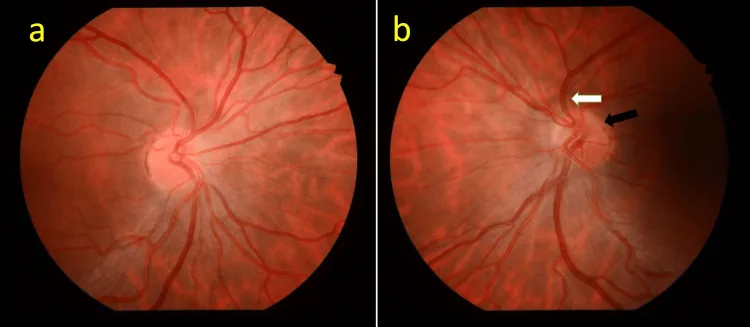
L’acuità visiva nell’ONH varia da normale a assenza di percezione luminosa. Nella maggior parte dei casi è inferiore a 0,1 e dipende dalla densità del fascio papillomaculare. L’ipoplasia del nervo ottico differisce da altre anomalie congenite della papilla in questo aspetto: anche se la macula è formata, il grado di sviluppo del fascio di fibre nervose papillomaculari varia, portando a un’ampia gamma di acuità visiva da 1,0 a molto bassa.
Secondo i dati giapponesi, una scarsa acuità visiva è stata osservata in 11/16 casi (69%), strabismo in 8/16 casi (50%) e nistagmo in 5/16 casi (31%)1).
All’oftalmoscopia si osservano reperti caratteristici.
I principali reperti oculari e la frequenza delle complicanze sistemiche sono mostrati di seguito.
| Reperto | Frequenza |
|---|---|
| Anomalie strutturali del SNC | Circa 90% |
| Disturbi del neurosviluppo | Circa 70% |
| Disfunzione ipotalamica (bilaterale) | 81% |
| Disfunzione ipotalamica (unilaterale) | 69% |
| Ritardo dello sviluppo (bilaterale) | 78% |
Anche in caso di unilateralità, la disfunzione ipotalamica è presente nel 69% dei casi e anomalie cerebrali sono presenti nel 18,2% dei pazienti asintomatici1). La RMN e lo screening endocrinologico sono obbligatori anche in caso di unilateralità.
La patologia dell’ONH è una disgenesia delle cellule gangliari retiniche (RGC) e delle fibre nervose. Esistono due ipotesi: la teoria della disgenesia e quella della degenerazione retrograda. È stata proposta anche un’ipotesi che considera l’ischemia del chiasma e del nervo ottico dovuta a disturbi vascolari dell’arteria cerebrale anteriore come eziologia.
La maggior parte dei casi è sporadica2)3).
Le seguenti mutazioni genetiche possono essere coinvolte.
La diagnosi di ONH si basa sui reperti oftalmoscopici e viene combinata con imaging e screening endocrinologico.
La conferma del segno del doppio anello è il primo passo diagnostico. Un rapporto DM/DD ≥ 3 (≥ 3,2 per papilla piccola) è un punto di riferimento.
In uno studio su pazienti giapponesi, sono state riscontrate anomalie cerebrali nel 43,8% dei casi e SOD nel 37,5%. È interessante notare che nel 18,2% dei pazienti asintomatici erano presenti anomalie cerebrali 1). Inoltre, in 2 dei 3 pazienti con ipofunzione ipofisaria, la RM mostrava una morfologia ipofisaria normale 1).
Per tutti i pazienti con ONH si raccomandano i seguenti esami.
È necessaria la diagnosi differenziale con le seguenti malattie.
Sì, è possibile. In uno studio su pazienti giapponesi, 2 dei 3 pazienti con insufficienza ipofisaria avevano una RM normale 1). Indipendentemente dai risultati della RM, lo screening endocrino dovrebbe essere eseguito in tutti i pazienti.
Non esiste un trattamento curativo per l’ONH stesso. La gestione si concentra sull’ottimizzazione della funzione visiva e sulla gestione delle complicanze sistemiche (in particolare le anomalie endocrine). È essenziale la collaborazione di un team multidisciplinare (oculistica, endocrinologia, pediatria, neurologia, riabilitazione). Si raccomanda una valutazione della crescita ogni sei mesi e della funzione visiva una volta all’anno.
Ormone della crescita
Frequenza : Necessario in circa il 70% dei pazienti con ONH.
Indicazione : Iniziare in caso di deficit confermato di ormone della crescita.
Ormone tiroideo
Frequenza : Necessario in circa il 43% dei pazienti.
Indicazione : Iniziare la sostituzione in caso di anomalie di TSH e FT4.
Ormone corticosteroideo (cortisolo)
Frequenza: necessaria in circa il 27% dei casi.
Attenzione: l’insufficienza surrenalica può essere fatale in situazioni di stress. L’istruzione sullo stress-dosing (aumento del dosaggio in caso di febbre/intervento chirurgico) è indispensabile 5).
Ormone antidiuretico
Frequenza: circa il 5% sviluppa diabete insipido.
Attenzione: una correzione rapida del sodio può causare convulsioni. La velocità di correzione deve essere inferiore a 0,5 mEq/L/h 4).
Nei casi adulti, sono stati riportati regimi sostitutivi come levotiroxina 137 µg, desmopressina, idrocortisone 10 mg (mattina)/7,5 mg (sera) 2).
L’ipoplasia del nervo ottico di per sé non è progressiva in assenza di glaucoma. In assenza di glaucoma, si devono evitare colliri o interventi chirurgici per abbassare la pressione intraoculare. Tuttavia, le anomalie endocrine possono comparire o peggiorare nel tempo, quindi è indispensabile un follow-up a lungo termine 1). Una diagnosi precoce e l’inizio della terapia ormonale sostitutiva entro i 3 anni di età nei casi necessari prevengono le sequele, quindi è importante tenere presente questa malattia anche nei casi unilaterali e non trascurarla.
L’ipoplasia del nervo ottico di per sé non è progressiva e, in assenza di glaucoma, la vista spesso rimane stabile. Tuttavia, le anomalie endocrine possono comparire successivamente, quindi è importante continuare una valutazione sistemica regolare.
L’essenza dell’ONH è una diminuzione dello strato di fibre nervose retiniche (RNFL) e delle cellule gangliari, con scarso effetto sugli strati esterni della retina. Esistono due ipotesi principali sul meccanismo di insorgenza.
Esiste anche l’ipotesi che l’ischemia della regione del chiasma ottico e del nervo ottico dovuta a un disturbo vascolare dell’arteria cerebrale anteriore sia coinvolta.
La disfunzione ipotalamica si osserva nel 69% dei casi di ONH unilaterale e nell’81% dei casi bilaterali. L’ipofisi e il nervo ottico sono vicini dal punto di vista dello sviluppo e si ritiene che lo stesso disturbo dello sviluppo colpisca entrambi.
Il ritardo dello sviluppo si osserva nel 75% di tutti i casi, con una frequenza maggiore nei casi bilaterali (78%) rispetto a quelli unilaterali (39%).
Il SOD plus è una condizione che, oltre al SOD classico, include displasie corticali (polimicrogiria, schizencefalia, ecc.) e secondo alcuni rapporti è più frequente del SOD classico7). La prognosi neuroevolutiva è peggiore e anche il rischio di epilessia è più elevato.
Come terapia farmacologica per l’obesità associata a SOD, è in corso uno studio clinico (NCT06760546) su setmelanotide, un agonista del recettore della melanocortina 4 (MC4R)2). L’obesità ipotalamica riduce significativamente la qualità della vita dei pazienti con SOD, pertanto è considerata una nuova opzione terapeutica.
Nei neonati SOD con glicemia normale, sono state riportate la gestione del micropene e del criptorchidismo con terapia a base di testosterone (25 mg IM, 1 volta al mese per 3 mesi) e tentativi di terapia con FSH ricombinante 5). Questi approcci mostrano il potenziale di un intervento precoce di sostituzione endocrina, ma l’efficacia e la sicurezza a lungo termine non sono ancora state stabilite.