O aconselhamento genético é um serviço médico cujo objetivo é fornecer informações genéticas corretas. Ele oferece aos pacientes e às famílias informações sobre o diagnóstico, o padrão de herança, o risco de recorrência, os exames disponíveis e as opções de tratamento das doenças genéticas, e apoia a tomada de decisões autônomas. Essa definição também é compartilhada internacionalmente, e os três pilares do aconselhamento genético são «fornecimento de informações», «apoio psicológico» e «apoio à tomada de decisões»3).
As doenças oculares hereditárias representam cerca de 43% da deficiência visual congênita1). A frequência de anomalias cromossômicas na descendência é de cerca de 0,5 a 1%. A deficiência de visão vermelho-verde é uma das doenças oculares hereditárias mais comuns, ocorrendo em cerca de 5% dos homens e 0,2% das mulheres. A prevalência da retinite pigmentosa é de cerca de 1/4.000 a 1/5.000, sendo uma das principais causas de deficiência visual2).
O aconselhamento genético não se destina apenas ao próprio paciente com a doença hereditária, mas também a familiares que possam desenvolvê-la e a portadores preocupados em transmiti-la a seus futuros filhos. O oftalmologista, ao mesmo tempo em que faz o diagnóstico, atua em conjunto com especialistas em genética e conselheiros genéticos certificados para fornecer informações.
QOnde o aconselhamento genético pode ser feito?
A
Pode ser feito nos departamentos de medicina genética de hospitais universitários ou hospitais de referência, ou em instituições com conselheiros genéticos certificados. É possível encontrar instituições consultando as listas de conselheiros genéticos certificados da Sociedade Japonesa de Aconselhamento Genético e da Sociedade Japonesa de Genética Humana. O site do Centro de Informações sobre Doenças Raras também fornece orientações sobre onde buscar atendimento.
2. Padrões de herança e principais doenças oculares hereditárias
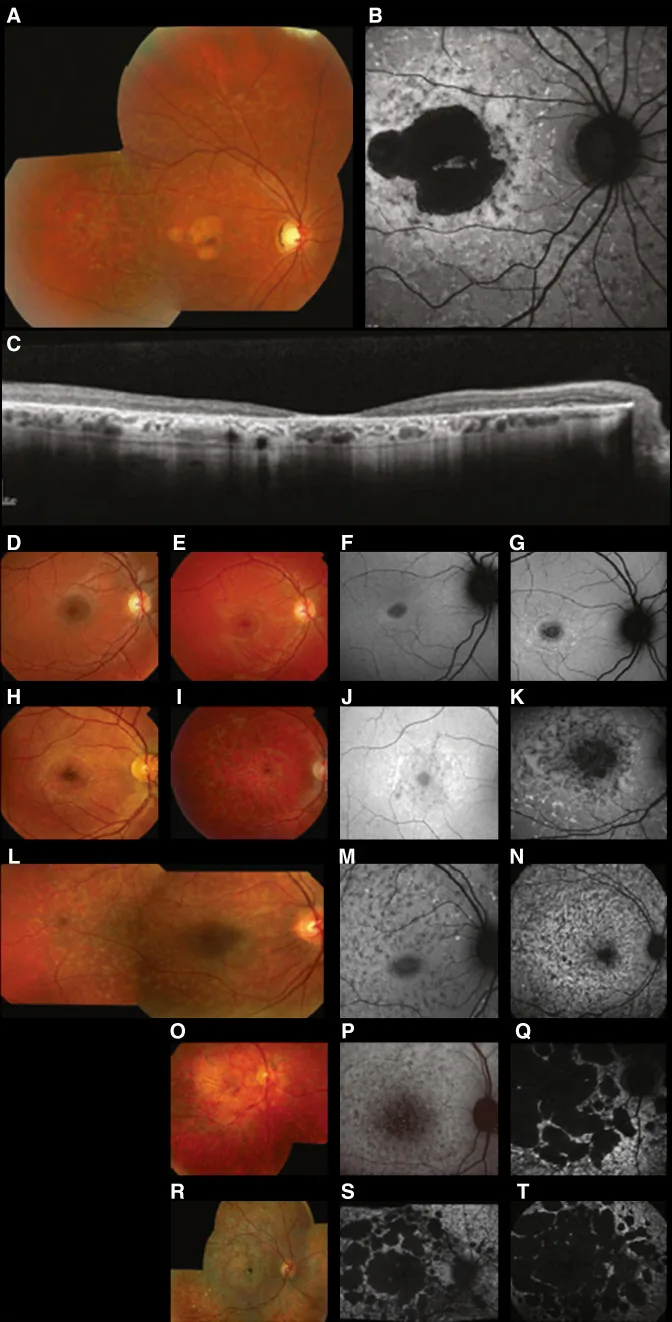
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Imagens multimodais típicas da doença de Stargardt (STGD1): a foto de fundo de olho em cores mostra manchas branco-amareladas ao nível do epitélio pigmentado da retina e atrofia macular (A); a autofluorescência do fundo de olho mostra uma área macular hipofluorescente com fluorescência anormal ao redor (B); e a SD-OCT mostra perda acentuada das camadas externas da retina e do EPR, com focos hiper-refletivos correspondentes às manchas (C). Corresponde à doença de Stargardt (uma distrofia retiniana autossômica recessiva causada por mutações no gene ABCA4) abordada na seção “2. Padrões de herança e principais doenças oculares hereditárias”.
O padrão de herança é uma informação central no aconselhamento genético e é classificado em quatro padrões principais.
Herança autossômica dominante
Condição de aparecimento: A doença ocorre com mutação em um alelo (estado heterozigoto).
Características da árvore genealógica: Os membros afetados aparecem em gerações consecutivas.
Risco de recorrência: A chance de transmitir para um filho é de 50%.
Observação: Se a penetrância não for de 100%, pode haver salto de geração.
Herança autossômica recessiva
Condição de aparecimento: A doença ocorre quando ambos os alelos estão mutados (homozigoto ou heterozigoto composto).
Características da árvore genealógica: Os membros afetados aparecem entre irmãos. Os pais geralmente são portadores (heterozigotos).
Risco de recorrência: A chance de doença em um filho de dois portadores é de 25%.
Tendência recente: Como os casamentos entre primos se tornaram menos comuns, a proporção de heterozigotos compostos aumentou.
Herança ligada ao X
Condição de manifestação: A maioria dos pacientes é do sexo masculino (hemizigoto).
Nas mulheres: Como têm dois cromossomos X, uma mutação em um deles as torna portadoras.
Características da árvore genealógica: Os afetados tendem a ser homens, e a transmissão ocorre da mãe para o filho.
Risco de recorrência: A probabilidade de um filho de mãe portadora ser afetado é de 50%.
Herança materna (herança mitocondrial)
Característica: O DNA mitocondrial (mtDNA) do espermatozoide é quase todo degradado na fecundação. Por isso, é transmitido apenas da mãe para o filho.
QSe um dos pais tiver uma doença ocular hereditária, qual é a chance de ela ser transmitida ao filho?
A
O padrão de herança varia. Na herança autossômica dominante, a chance de transmissão de um dos pais afetado para o filho é de 50%. Na herança autossômica recessiva, se ambos os pais forem portadores, a chance de o filho desenvolver a doença é de 25%. Na herança ligada ao cromossomo X, a chance de um filho homem de uma mãe portadora ser afetado é de 50%, e na herança materna (mitocondrial), se a mãe estiver afetada ou for portadora, ela pode ser transmitida a todos os filhos. Para situações individuais, recomenda-se consultar um geneticista ou um conselheiro genético certificado.
3. Causas e fatores de risco das doenças oculares hereditárias
Os mecanismos de aparecimento das doenças oculares hereditárias são classificados de acordo com o tipo de efeito que uma mutação genética exerce sobre a função das proteínas.
Haploinsuficiência (haploinsufficiency): O fenótipo surge quando a função de apenas um alelo é perdida. Um exemplo clássico é a aniridia congênita (mutação de PAX6). Um único alelo normal não consegue fornecer a quantidade de produto gênico necessária para a formação normal dos tecidos.
Efeito negativo dominante (dominant negative effect): A proteína mutante inibe a função da proteína normal de forma competitiva e estrutural. Na síndrome de Marfan (mutação de FBN1), moléculas de fibrilina-1 mutante prejudicam a formação da matriz extracelular.
Mutações mitocondriais: Na neuropatia óptica hereditária de Leber (LHON), três mutações pontuais — 11778, 3460 e 14484 — correspondem a cerca de 90% de todas as mutações. A produção de energia prejudicada danifica as células ganglionares da retina.
Mutações de novo: Mutações que não estão presentes nos pais e surgem novo durante a formação do óvulo ou do espermatozoide. Um exemplo representativo é a amaurose congênita de Leber causada por mutações em CRX. Também podem ocorrer como casos isolados quando ninguém mais na árvore genealógica é afetado.
Heterozigoto composto: Em doenças autossômicas recessivas, é a forma em que ocorrem duas mutações diferentes em dois alelos. Com a recente diminuição dos casamentos entre primos, a proporção de heterozigotos compostos aumentou em comparação com os homozigotos.
Dissomia uniparental: Fenômeno em que as duas cópias do mesmo cromossomo são herdadas de um dos pais, enquanto o cromossomo do outro pai está ausente. Pode aparecer como um caso aparentemente esporádico10).
Mais de 100 genes causadores de retinite pigmentosa já foram identificados, e até o mesmo fenótipo pode ser causado por muitos genes diferentes2). Essa diversidade genética torna o diagnóstico difícil.
4. Aspectos práticos do aconselhamento genético e do teste genético
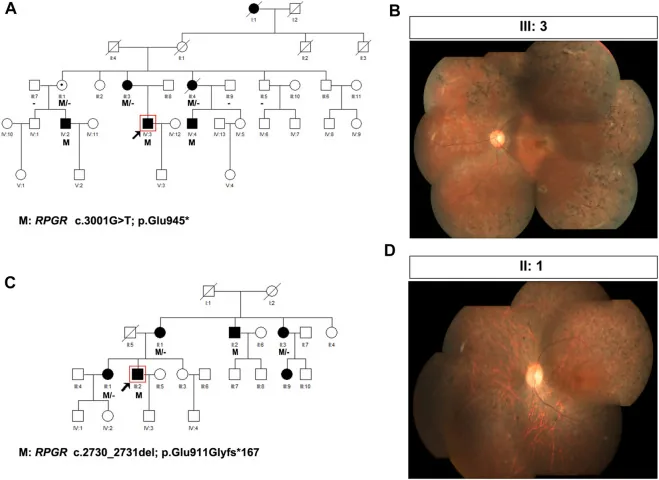
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Pedigrees e fotografias de fundo de olho de duas famílias com retinose pigmentar ligada ao X (variantes de RPGR: c.3001G>T e c.2730_2731del). Os pedigrees usam símbolos padrão (quadrados preenchidos para homens afetados, círculos pontilhados para mulheres portadoras) para mostrar o padrão de herança, e as fotografias de fundo de olho correspondentes mostram pigmentação em espículas ósseas avançada e atrofia da retina. Isso corresponde à elaboração do pedigree e à determinação do padrão de herança discutidas na seção “4. A prática do aconselhamento genético e do teste genético”.
Para realizar o aconselhamento genético de forma adequada, é necessário o seguinte preparo.
Elaboração de um pedigree: Obter histórico familiar de pelo menos três gerações e registrá-lo como pedigree. A distribuição vertical e horizontal dos afetados permite estimar o padrão de herança.
Estimativa do padrão de herança: A partir do pedigree e do fenótipo, inferir se é autossômico dominante, autossômico recessivo, ligado ao X ou materno.
Explicação do teste genético e obtenção de consentimento por escrito: Antes de examinar as informações genéticas do paciente, é necessário explicar bem o significado e os problemas do teste e obter consentimento por escrito.
Anonimização dos resultados do teste de DNA e gestão das informações: Proteger a privacidade por meio de anonimização com possibilidade de vinculação.
Definição prévia da conduta para achados incidentais: Confirmar com o paciente, antes do teste, a política de comunicação caso sejam detectados achados incidentais graves que possam ameaçar a vida.
Alta precisão. Adequado para confirmar locais de mutação já conhecidos
Teste em painel (sequenciamento direcionado)
Grupo de genes relacionados a doenças específicas
Pesquisa em conjunto de genes relacionados a doenças da retina. Alta taxa de diagnóstico5)
Análise do exoma (NGS)
Todas as regiões exônicas
Detecta variantes desconhecidas. Útil quando há muitos genes causadores da doença
Análise do genoma completo (WGS)
Genoma inteiro
Pode ter uma taxa de diagnóstico mais alta do que os testes NGS direcionados existentes13)
Na distrofia retinal hereditária, quando se suspeita de uma doença causada por uma mutação no gene RPE65, e em casos como o de ajudar a determinar a indicação para terapia gênica, alguns exames que atendem aos critérios podem ser realizados como atendimento coberto pelo seguro4).
Interpretação das variantes e principais bases de dados
A interpretação das variantes exige julgamento cuidadoso. Sabe-se que cerca de 30% das descrições publicadas como “variantes” em artigos são, na verdade, polimorfismos (variantes normais). Em geral, alterações na sequência de bases que aparecem em 1 ou mais de cada 100 pessoas saudáveis devem ser tratadas como polimorfismos.
As seguintes bases de dados principais são utilizadas.
OMIM (Online Mendelian Inheritance in Man): base de dados abrangente de doenças e genes hereditários
GeneReviews: fornece informações de aconselhamento genético para cada doença
RetNet (Retinal Information Network): base de dados genética especializada em doenças da retina
QO custo do teste genético é coberto pelo seguro?
A
Em algumas doenças oculares hereditárias, o teste genético pode ser realizado como atendimento coberto pelo seguro quando a indicação terapêutica é determinada e os critérios da instituição designada são atendidos. No entanto, a cobertura depende do tipo de exame e da doença, por isso é necessário confirmar na instituição onde você será atendido. Exames não cobertos pelo seguro podem ser pagos pelo próprio paciente. Em doenças designadas como raras e de difícil tratamento (como a retinose pigmentar), também pode haver apoio financeiro por meio do sistema de subsídio para despesas médicas de doenças raras e de difícil tratamento4).
5. Sistema de aconselhamento genético e perspectivas de tratamento
Na realização do aconselhamento genético, a colaboração com um médico especialista em genética e um conselheiro genético certificado é considerada ideal. Em hospitais universitários, pode ser necessária a revisão por um comitê de ética. Como a informação genética pode afetar não apenas o paciente, mas também os familiares, é necessário cuidado especial no seu manejo. O debate social sobre a prevenção da discriminação injusta com base em informações genéticas também está avançando8).
Doenças raras designadas e auxílio com despesas médicas
Em pacientes com doenças raras, o copagamento médico é reduzido. É estabelecido um teto mensal para o total de consultas ambulatoriais, internações e dispensação de medicamentos, e são aplicadas categorias de acordo com a renda.
O medicamento de terapia gênica voretigene neparvovec para distrofia retiniana hereditária causada por mutações no gene RPE65 é uma formulação para administração sub-retiniana que utiliza um vetor AAV2. Sua eficácia e segurança foram confirmadas em ensaios randomizados controlados6). A decisão sobre a indicação do tratamento requer a confirmação do tipo da doença por teste genético.
Quanto à terapia com oligonucleotídeos antisense (ASO), a administração intravítrea de uma formulação para amaurose congênita de Leber tipo 10 (LCA10) causada por mutações em CEP290 está sendo estudada em ensaios clínicos9).
O transplante autólogo de células do epitélio pigmentar da retina usando células iPS vem sendo estudado como medicina regenerativa para degeneração macular relacionada à idade7). Embora seja uma área diferente das doenças retinianas hereditárias, chama atenção como exemplo real de terapia celular da retina.
QA terapia génica já está disponível?
A
Na distrofia retiniana hereditária causada por mutação em RPE65, a eficácia e a segurança de voretigene neparvovec foram demonstradas em ECRs6). Para confirmar se o tratamento está indicado, é necessário definir o tipo de doença por exame genético. Na LCA10 com mutação em CEP290, a injeção intravítrea de formulações ASO está a ser estudada em ensaios clínicos9).
A herança autossómica dominante, autossómica recessiva e ligada ao X segue as leis de Mendel. No entanto, os seguintes fatores podem dificultar uma previsão simples.
Penetrância: mesmo que a pessoa tenha a mutação, nem todas desenvolvem a doença. Quando a penetrância é baixa, a alteração pode saltar gerações, tornando difícil inferir o padrão de herança a partir da árvore genealógica.
Expressividade: mesmo entre familiares com o mesmo gene mutado, a gravidade dos sintomas pode variar.
Mutação de ganho de função: mecanismo em que a proteína mutada adquire uma nova função prejudicial. Isto é diferente do efeito dominante negativo habitual.
As mitocôndrias נמצam-se no citoplasma, e apenas o mtDNA proveniente do óvulo da mãe é transmitido ao filho. Em cada célula existem milhares de cópias de mtDNA, e pode ocorrer um estado em que mtDNA mutado e mtDNA normal coexistem (heteroplasmia). Quanto maior a proporção de heteroplasmia, mais graves tendem a ser os sintomas. Na neuropatia óptica hereditária de Leber (LHON), três variantes—11778 (a mais frequente), 3460 e 14484—respondem por cerca de 90% de todas as variantes.
A dissomia uniparental é uma situação em que os dois cromossomos de um par vêm do mesmo progenitor, sem cromossomo do outro progenitor. Um filho de um portador de doença autossômica recessiva pode desenvolver a doença mesmo que o outro progenitor não seja portador, criando um caso esporádico aparente10). Junto com variantes de novo e heterozigotos compostos, a doença hereditária deve ser considerada mesmo quando não há histórico familiar.
Na LCA10 com variantes de CEP290, a correção de splicing por terapia ASO está sendo estudada em ensaios clínicos9).
O candidato a terapia de edição gênica EDIT-101, usando CRISPR/Cas9, foi relatado em desenvolvimento pré-clínico para LCA10 (variantes de CEP290)11).
O teste genético pré-implantacional (PGT-M) pode ser realizado para doenças hereditárias autossômicas dominantes e recessivas, e pode ser considerado dentro de uma estrutura ética12).
A sequenciação do genoma completo (WGS) mostrou potencial para aumentar a taxa de diagnóstico molecular em doenças hereditárias da retina em comparação com os testes genéticos padrão existentes13).
Ferramentas de previsão de patogenicidade de variantes genéticas com uso de inteligência artificial (IA) estão sendo desenvolvidas, e espera-se melhorar a precisão da interpretação das variantes.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.