Le conseil génétique est un service médical dont l’objectif est de fournir une information génétique exacte. Il donne aux patients et à leur famille des informations sur le diagnostic, le mode de transmission, le risque de récidive, les examens disponibles et les traitements des maladies génétiques, et soutient une prise de décision autonome. Cette définition est également partagée au niveau international, et les trois piliers du conseil génétique sont la « fourniture d’informations », le « soutien psychologique » et le « soutien à la prise de décision »3).
Les maladies oculaires héréditaires représentent environ 43 % des déficiences visuelles congénitales1). La fréquence des anomalies chromosomiques chez la descendance est d’environ 0,5 à 1 %. Le trouble de la vision rouge-vert est l’une des maladies oculaires héréditaires les plus fréquentes, touchant environ 5 % des hommes et 0,2 % des femmes. La prévalence de la rétinite pigmentaire est d’environ 1/4 000 à 1/5 000, ce qui en fait l’une des principales causes de déficience visuelle2).
Le conseil génétique ne concerne pas seulement le patient atteint de la maladie héréditaire, mais aussi les membres de la famille susceptibles de la développer et les porteurs qui s’inquiètent de la transmettre à leurs futurs enfants. L’ophtalmologiste, tout en assurant le diagnostic, travaille avec des spécialistes en génétique et des conseillers en génétique certifiés pour fournir des informations.
QOù peut-on recevoir un conseil génétique ?
A
Il peut être reçu dans les services de génétique des hôpitaux universitaires ou des hôpitaux de référence, ou dans des établissements où exercent des conseillers en génétique certifiés. On peut trouver ces établissements en consultant les listes de conseillers en génétique certifiés de la Société japonaise de conseil génétique et de la Société japonaise de génétique humaine. Le site du Centre d’information sur les maladies rares fournit également des indications sur les lieux de consultation.
2. Modes de transmission et principales maladies oculaires héréditaires
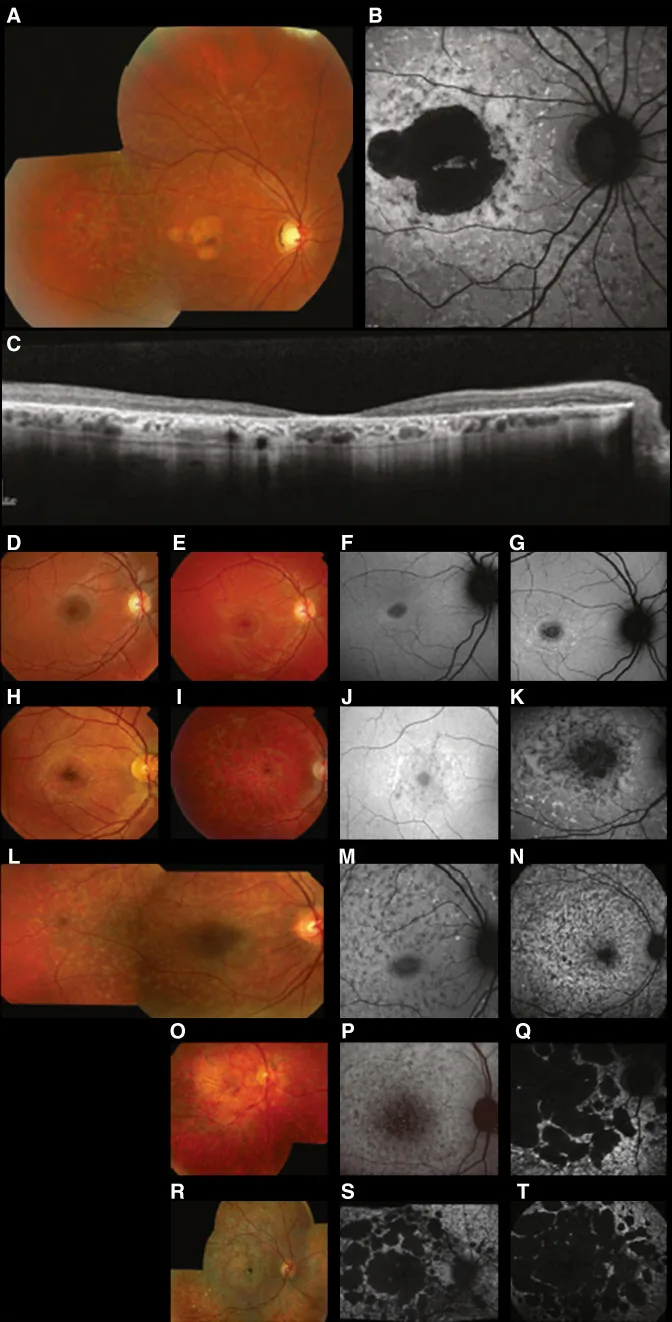
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Imagerie multimodale typique de la maladie de Stargardt (STGD1) : la photographie couleur du fond d’œil montre des plages jaune-blanchâtres au niveau de l’épithélium pigmentaire rétinien et une atrophie maculaire (A) ; l’autofluorescence du fond d’œil montre une zone maculaire hypoautofluorescente avec une fluorescence anormale autour (B) ; et la SD-OCT montre une disparition marquée des couches externes de la rétine et de l’EPR, avec des foyers hyperréflectifs correspondant aux plages (C). Cela correspond à la maladie de Stargardt (une dystrophie rétinienne autosomique récessive causée par des mutations du gène ABCA4) abordée dans la section « 2. Modes de transmission et principales maladies oculaires héréditaires ».
Le mode de transmission est une information essentielle du conseil génétique et se classe en quatre grands modes.
Hérédité autosomique dominante
Condition d’apparition : La maladie survient avec une mutation sur un seul allèle (état hétérozygote).
Caractéristiques de l’arbre généalogique : Les membres atteints apparaissent à des générations successives.
Risque de récidive : La probabilité de transmission à un enfant est de 50%.
Remarque : Si la pénétrance n’est pas de 100%, une génération peut être sautée.
Hérédité autosomique récessive
Condition d’apparition : La maladie survient lorsque les deux allèles sont mutés (homozygote ou hétérozygote composite).
Caractéristiques de l’arbre généalogique : Les membres atteints apparaissent entre frères et sœurs. Les parents sont généralement porteurs (hétérozygotes).
Risque de récidive : La probabilité de maladie chez un enfant de deux porteurs est de 25%.
Tendance récente : Comme les mariages entre cousins sont moins fréquents, la proportion d’hétérozygotes composites a augmenté.
Hérédité liée à l’X
Condition d’apparition : La plupart des patients sont des hommes (hémizygotes).
Chez les femmes : Comme elles ont deux chromosomes X, une mutation sur l’un d’eux les rend porteuses.
Caractéristiques de l’arbre généalogique : Les personnes atteintes sont surtout des hommes, et la transmission se fait de la mère au fils.
Risque de récurrence : La probabilité qu’un fils d’une mère porteuse soit atteint est de 50 %.
Hérédité maternelle (hérédité mitochondriale)
Caractéristique : L’ADN mitochondrial (mtDNA) du spermatozoïde est presque entièrement dégradé lors de la fécondation. Il est donc transmis uniquement par la mère à l’enfant.
QSi l’un des parents a une maladie oculaire héréditaire, quelle est la probabilité de la transmettre à l’enfant ?
A
Le mode de transmission varie. Dans l’hérédité autosomique dominante, la probabilité de transmission d’un parent atteint à un enfant est de 50 %. Dans l’hérédité autosomique récessive, si les deux parents sont porteurs, la probabilité qu’un enfant développe la maladie est de 25 %. Dans l’hérédité liée à l’X, la probabilité qu’un fils d’une mère porteuse soit atteint est de 50 %, et dans l’hérédité maternelle (mitochondriale), si la mère est atteinte ou porteuse, la transmission peut concerner tous les enfants. Pour chaque situation, il est recommandé de consulter un spécialiste en génétique ou un conseiller en génétique certifié.
3. Causes et facteurs de risque des maladies oculaires héréditaires
Les mécanismes d’apparition des maladies oculaires héréditaires sont classés selon le type d’effet qu’une mutation génétique exerce sur la fonction des protéines.
Haplo-insuffisance (haploinsufficiency) : le phénotype apparaît lorsque la fonction d’un seul allèle est perdue. L’exemple classique est l’aniridie congénitale (mutation de PAX6). Un seul allèle normal ne peut pas fournir la quantité de produit génique nécessaire à la formation normale des tissus.
Effet dominant négatif (dominant negative effect) : la protéine mutée inhibe la fonction de la protéine normale de manière compétitive et structurale. Dans le syndrome de Marfan (mutation de FBN1), les molécules de fibrilline-1 mutées perturbent la formation de la matrice extracellulaire.
Mutations mitochondriales : Dans la neuropathie optique héréditaire de Leber (LHON), trois mutations ponctuelles — 11778, 3460 et 14484 — représentent environ 90 % de toutes les mutations. Le trouble de la production d’énergie endommage les cellules ganglionnaires rétiniennes.
Mutations de novo : Mutations absentes chez les parents et apparaissant de novo lors de la formation de l’ovule ou du spermatozoïde. Un exemple représentatif est l’amaurose congénitale de Leber causée par des mutations de CRX. Elles peuvent aussi survenir comme cas isolés lorsque personne d’autre dans l’arbre familial n’est atteint.
Hétérozygote composite : dans les maladies autosomiques récessives, il s’agit d’une forme où deux mutations différentes surviennent sur les deux allèles. Avec la récente diminution des mariages entre cousins, la proportion d’hétérozygotes composites a augmenté par rapport aux homozygotes.
Disomie uniparentale : phénomène dans lequel les deux copies du même chromosome sont héritées d’un seul parent, tandis que le chromosome de l’autre parent est absent. Elle peut se présenter comme un cas apparemment sporadique10).
Plus de 100 gènes responsables de la rétinite pigmentaire ont été identifiés, et un même phénotype peut même être causé par de nombreux gènes différents2). Cette diversité génétique rend le diagnostic difficile.
4. Aspects pratiques du conseil génétique et des tests génétiques
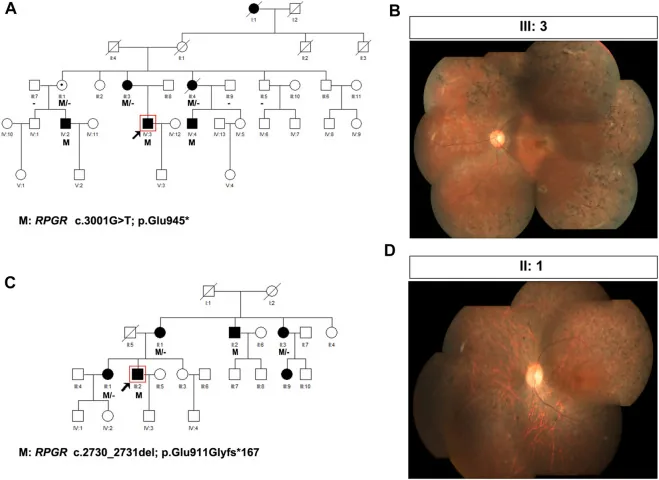
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Arbres généalogiques et photographies du fond d’œil de deux familles atteintes de rétinite pigmentaire liée à l’X (variants de RPGR : c.3001G>T et c.2730_2731del). Les arbres généalogiques utilisent des symboles standard (carrés pleins pour les hommes atteints, cercles pointillés pour les femmes porteuses) pour montrer le mode de transmission, et les photographies du fond d’œil correspondantes montrent une pigmentation avancée en spicules osseux et une atrophie rétinienne. Cela correspond à la réalisation de l’arbre généalogique (pedigree) et à la détermination du mode de transmission abordées dans la section « 4. Pratique du conseil génétique et des tests génétiques ».
Pour réaliser correctement le conseil génétique, la préparation suivante est nécessaire.
Création d’un arbre généalogique (pedigree) : Recueillir les antécédents familiaux sur au moins trois générations et les consigner sous forme d’arbre généalogique. La répartition verticale et horizontale des personnes atteintes permet d’estimer le mode de transmission.
Estimation du mode de transmission : À partir de l’arbre généalogique et du phénotype, déterminer s’il s’agit d’une transmission autosomique dominante, autosomique récessive, liée à l’X ou maternelle.
Explication du test génétique et obtention du consentement écrit : Avant d’examiner les informations génétiques du patient, il faut expliquer suffisamment la signification et les limites du test, puis obtenir un consentement écrit.
Anonymisation des résultats du test ADN et gestion des informations : Protéger la vie privée grâce à une anonymisation réversible.
Définition préalable de la conduite à tenir en cas de découverte fortuite : Confirmer avec le patient, avant le test, la politique de communication si des découvertes fortuites graves susceptibles d’engager le pronostic vital sont mises en évidence.
Très précis. Adapté à la confirmation de sites de mutation connus
Test en panel (séquençage ciblé)
Groupe de gènes associés à des maladies spécifiques
Recherche simultanée des gènes liés aux maladies de la rétine. Taux de diagnostic élevé5)
Analyse de l’exome (NGS)
Toutes les régions exoniques
Détecte les variants inconnus. Utile lorsqu’il existe de nombreux gènes responsables d’une maladie
Analyse du génome entier (WGS)
Génome entier
Peut avoir un taux de diagnostic plus élevé que les tests NGS ciblés existants13)
Dans la dystrophie rétinienne héréditaire, lorsqu’une maladie due à une mutation du gène RPE65 est suspectée, et dans des cas tels que l’aide à la décision d’une indication de thérapie génique, certains examens remplissant les conditions peuvent être réalisés dans le cadre des soins pris en charge par l’assurance4).
Interprétation des variants et principales bases de données
L’interprétation des variants nécessite un jugement prudent. On sait qu’environ 30 % des descriptions publiées comme « variants » dans les articles sont en réalité des polymorphismes (variants normaux). En général, les modifications de séquence présentes chez une personne saine ou plus sur 100 doivent être considérées comme des polymorphismes.
Les principales bases de données suivantes sont utilisées.
OMIM (Online Mendelian Inheritance in Man) : base de données complète sur les maladies et les gènes héréditaires
GeneReviews : fournit des informations de conseil génétique pour chaque maladie
RetNet (Retinal Information Network) : base de données de gènes spécialisée dans les maladies rétiniennes
QLes frais de test génétique sont-ils pris en charge par l’assurance ?
A
Pour certaines maladies oculaires héréditaires, les tests génétiques peuvent être réalisés dans le cadre de soins pris en charge par l’assurance lorsque l’indication thérapeutique est déterminée et que les critères de l’établissement désigné sont remplis. Toutefois, la prise en charge dépend du type de test et de la maladie concernée, il faut donc vérifier auprès de l’établissement consulté. Les tests non couverts par l’assurance peuvent être à la charge du patient. Pour les maladies désignées comme incurables (par exemple la rétinite pigmentaire), un soutien financier peut également être disponible via le système de subvention des frais médicaux pour les maladies incurables désignées4).
5. Système de conseil génétique et perspectives de traitement
Lors de la mise en place d’un conseil génétique, une collaboration avec un médecin spécialiste en génétique et un conseiller en génétique certifié est considérée comme idéale. Dans les hôpitaux universitaires, un examen par un comité d’éthique peut être nécessaire. Comme l’information génétique peut affecter non seulement le patient mais aussi les membres de sa famille, une attention particulière est requise dans sa gestion. Les débats de société sur la prévention des discriminations injustes fondées sur les informations génétiques progressent également8).
Maladies rares désignées et aide aux frais médicaux
Chez les patients atteints de maladies rares, le reste à charge des frais médicaux est réduit. Un plafond mensuel est fixé pour le total des soins ambulatoires, des hospitalisations et des médicaments délivrés, et des catégories selon le revenu sont appliquées.
Le médicament de thérapie génique voretigene neparvovec pour la dystrophie rétinienne héréditaire causée par des mutations du gène RPE65 est une préparation administrée par voie sous-rétinienne utilisant un vecteur AAV2. Son efficacité et sa sécurité ont été confirmées dans des essais randomisés contrôlés6). La décision d’indiquer le traitement nécessite la confirmation du type de maladie par test génétique.
Pour la thérapie par oligonucléotides antisens (ASO), l’administration intravitréenne d’une préparation destinée à l’amaurose congénitale de Leber de type 10 (LCA10) causée par des mutations de CEP290 est étudiée dans des essais cliniques9).
La transplantation autologue de cellules de l’épithélium pigmentaire rétinien à l’aide de cellules iPS fait l’objet de recherches comme médecine régénérative pour la dégénérescence maculaire liée à l’âge7). Bien qu’il s’agisse d’un domaine différent des maladies rétiniennes héréditaires, cela attire l’attention comme exemple concret de thérapie cellulaire rétinienne.
Dans la dystrophie rétinienne héréditaire due à une mutation de RPE65, l’efficacité et la sécurité du voretigene neparvovec ont été démontrées dans des ECR6). Pour confirmer l’indication du traitement, il faut préciser le type de maladie par un test génétique. Dans la LCA10 associée à une mutation de CEP290, l’injection intravitréenne de préparations d’ASO est étudiée dans des essais cliniques9).
L’hérédité autosomique dominante, autosomique récessive et liée à l’X suit les lois de Mendel. Cependant, les facteurs suivants peuvent rendre la prédiction simple difficile.
Pénétrance : même en présence de la mutation, toutes les personnes ne développent pas la maladie. Lorsque la pénétrance est faible, le trouble peut sauter des générations, ce qui rend difficile l’inférence du mode de transmission à partir de l’arbre généalogique.
Expressivité : même au sein d’une famille portant le même gène muté, la sévérité des symptômes peut varier.
Mutation gain de fonction : mécanisme dans lequel la protéine mutée acquiert une nouvelle fonction nocive. Cela diffère de l’effet dominant négatif habituel.
Les mitochondries se trouvent dans le cytoplasme, et seul l’ADN mitochondrial (mtDNA) provenant de l’ovule de la mère est transmis à l’enfant. Chaque cellule contient des milliers de copies de mtDNA, et un état où du mtDNA muté et du mtDNA normal coexistent (hétéroplasmie) peut apparaître. Plus la proportion d’hétéroplasmie est élevée, plus les symptômes ont tendance à être sévères. Dans la neuropathie optique héréditaire de Leber (LHON), trois variants—11778 (le plus fréquent), 3460 et 14484—représentent environ 90 % de l’ensemble des variants.
La disomie uniparentale est une situation dans laquelle les deux chromosomes d’une paire proviennent du même parent, sans chromosome de l’autre parent. Un enfant né d’un porteur d’une maladie autosomique récessive peut développer la maladie même si l’autre parent n’est pas porteur, donnant l’apparence d’un cas sporadique10). Avec les variants de novo et les hétérozygotes composites, une maladie héréditaire doit être envisagée même en l’absence d’antécédents familiaux.
7. Recherches les plus récentes et perspectives d’avenir
Dans la LCA10 avec variants de CEP290, la correction de l’épissage par thérapie ASO fait l’objet d’études dans des essais cliniques9).
Le candidat EDIT-101, une thérapie d’édition du génome utilisant CRISPR/Cas9, a été signalé en développement préclinique pour la LCA10 (variants de CEP290)11).
Le diagnostic génétique préimplantatoire (PGT-M) peut être réalisé pour les maladies héréditaires autosomiques dominantes et récessives, et peut parfois être envisagé dans un cadre éthique12).
Le séquençage du génome entier (WGS) a montré qu’il pouvait augmenter le taux de diagnostic moléculaire dans les maladies rétiniennes héréditaires par rapport aux tests génétiques standards existants13).
Des outils d’IA pour prédire la pathogénicité des variants génétiques sont en cours de développement, et une amélioration de la précision de l’interprétation des variants est attendue.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.