Konseling genetik adalah layanan medis yang bertujuan untuk memberikan informasi genetik yang benar. Layanan ini memberikan informasi kepada pasien dan keluarga tentang diagnosis, pola pewarisan, risiko kekambuhan, pemeriksaan yang tersedia, dan pilihan pengobatan untuk penyakit genetik, serta mendukung pengambilan keputusan secara mandiri. Definisi ini juga diakui secara internasional, dan tiga pilar konseling genetik adalah «pemberian informasi», «dukungan psikologis», dan «dukungan pengambilan keputusan»3).
Penyakit mata herediter mencakup sekitar 43% dari gangguan penglihatan kongenital1). Frekuensi kelainan kromosom pada keturunan sekitar 0,5–1%. Gangguan penglihatan warna merah-hijau adalah salah satu penyakit mata herediter yang paling umum, muncul pada sekitar 5% laki-laki dan 0,2% perempuan. Prevalensi retinitis pigmentosa sekitar 1/4.000–1/5.000, dan merupakan salah satu penyebab utama gangguan penglihatan2).
Sasaran konseling genetik bukan hanya pasien dengan penyakit herediter itu sendiri, tetapi juga anggota keluarga yang mungkin mengalaminya dan pembawa yang khawatir akan mewariskannya kepada anak di masa depan. Dokter mata berperan dalam menegakkan diagnosis sambil bekerja sama dengan dokter spesialis genetika dan konselor genetik bersertifikat untuk memberikan informasi.
QDi mana konseling genetik dapat diperoleh?
A
Dapat diperoleh di bagian layanan genetika rumah sakit universitas atau rumah sakit inti, atau di fasilitas yang memiliki konselor genetik bersertifikat. Fasilitas dapat dicari dengan merujuk pada daftar konselor genetik bersertifikat dari Japanese Society of Genetic Counseling dan Japanese Society of Human Genetics. Situs web Nanbyo Information Center juga menyediakan panduan tempat berkonsultasi.
2. Pola pewarisan dan penyakit mata herediter utama
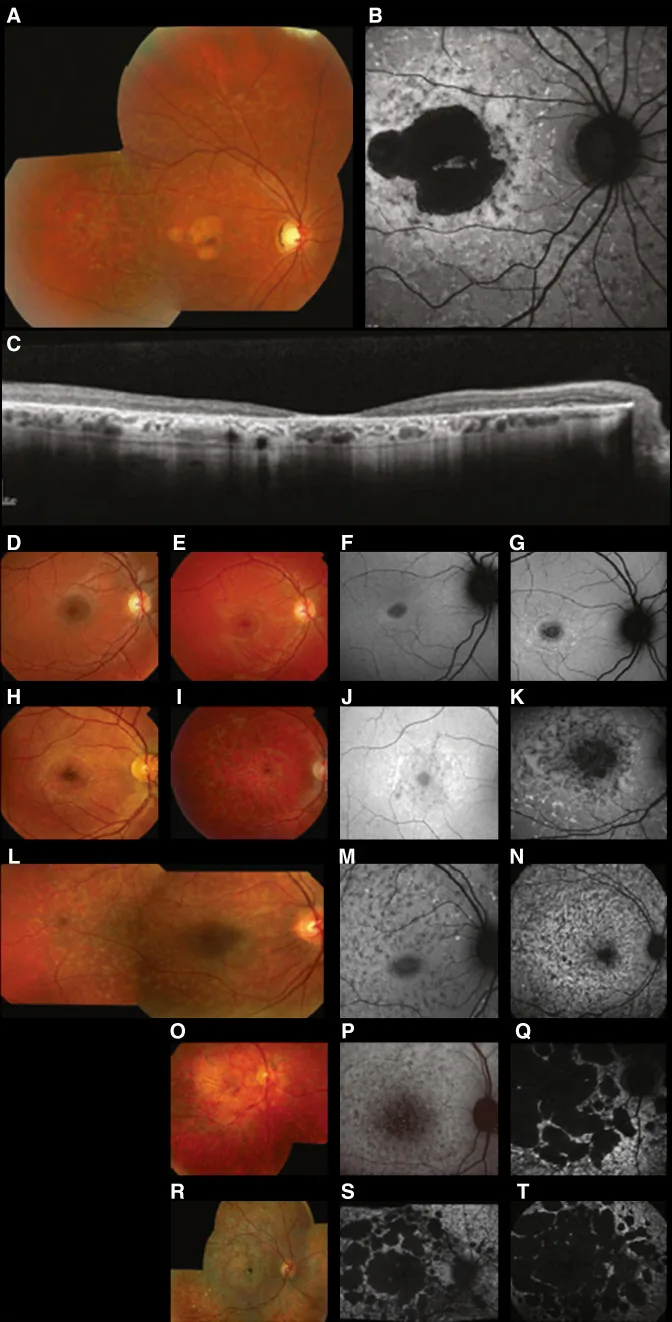
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Citra multimodal khas penyakit Stargardt (STGD1): foto fundus berwarna menunjukkan bercak putih kekuningan pada tingkat epitel pigmen retina dan atrofi makula (A); autofluoresensi fundus menunjukkan area hipofluoresen di makula dengan fluoresensi abnormal di sekitarnya (B); dan SD-OCT menunjukkan hilangnya lapisan retina luar dan RPE secara nyata, dengan fokus hiperreflektif yang sesuai dengan bercak tersebut (C). Ini sesuai dengan penyakit Stargardt (distrofi retina resesif autosomal akibat mutasi gen ABCA4) yang dibahas dalam bagian “2. Pola pewarisan dan penyakit mata herediter utama”.
Pola pewarisan merupakan informasi inti dalam konseling genetik dan dibagi menjadi empat pola utama.
Pewarisan autosomal dominan
Kondisi muncul: Penyakit terjadi dengan mutasi pada satu alel (keadaan heterozigot).
Ciri pohon keluarga: Anggota yang terkena muncul pada generasi yang berurutan.
Risiko kekambuhan: Kemungkinan diturunkan ke anak adalah 50%.
Catatan: Jika penetrans tidak 100%, satu generasi dapat terlewat.
Pewarisan autosomal resesif
Kondisi muncul: Penyakit terjadi ketika kedua alel bermutasi (homozigot atau heterozigot majemuk).
Ciri pohon keluarga: Anggota yang terkena muncul di antara saudara kandung. Orang tua biasanya pembawa (heterozigot).
Risiko kekambuhan: Kemungkinan penyakit pada anak dari dua pembawa adalah 25%.
Tren terbaru: Karena pernikahan antarsepupu makin jarang, proporsi heterozigot majemuk meningkat.
Pewarisan terpaut X
Kondisi terjadinya: Sebagian besar pasien adalah laki-laki (hemizigot).
Pada perempuan: Karena memiliki dua kromosom X, bila satu mengalami mutasi maka menjadi pembawa.
Ciri silsilah: Penderita lebih banyak pada laki-laki, dan diturunkan dari ibu ke anak laki-laki.
Risiko kekambuhan: Kemungkinan putra dari ibu pembawa untuk mengalami penyakit adalah 50%.
Pewarisan maternal (pewarisan mitokondria)
Ciri: DNA mitokondria (mtDNA) pada sperma hampir seluruhnya diuraikan saat pembuahan. Karena itu, penularannya hanya dari ibu ke anak.
QJika salah satu orang tua memiliki penyakit mata herediter, berapa kemungkinan diturunkan ke anak?
A
Pola pewarisan berbeda. Pada pewarisan autosomal dominan, peluang menurunkannya dari orang tua yang terkena ke anak adalah 50%. Pada pewarisan autosomal resesif, jika kedua orang tua adalah pembawa, peluang anak terkena penyakit adalah 25%. Pada pewarisan terpaut kromosom X, peluang anak laki-laki dari ibu pembawa untuk terkena adalah 50%, dan pada pewarisan maternal (mitokondria), jika ibu terkena atau menjadi pembawa, penyakit dapat diteruskan ke semua anak. Untuk situasi masing-masing, disarankan berkonsultasi dengan dokter spesialis genetika atau konselor genetik bersertifikat.
3. Penyebab dan faktor risiko penyakit mata herediter
Mekanisme terjadinya penyakit mata herediter diklasifikasikan menurut jenis dampak variasi genetik terhadap fungsi protein.
Haploinsufisiensi (haploinsufficiency): Fenotipe muncul hanya dengan hilangnya fungsi satu alel. Contoh khasnya adalah aniridia kongenital (mutasi PAX6). Satu alel normal tidak cukup untuk menyediakan jumlah produk gen yang diperlukan untuk pembentukan jaringan normal.
Efek negatif dominan (dominant negative effect): Protein mutan menghambat fungsi protein normal secara kompetitif dan struktural. Pada sindrom Marfan (mutasi FBN1), molekul fibrillin-1 mutan mengganggu pembentukan matriks ekstraseluler.
Mutasi mitokondria: Pada neuropati optik herediter Leber (LHON), tiga mutasi titik—11778, 3460, dan 14484—menyumbang sekitar 90% dari seluruh mutasi. Gangguan produksi energi merusak sel ganglion retina.
Mutasi de novo: Mutasi yang tidak ada pada orang tua dan muncul baru saat pembentukan sel telur atau sperma. Contoh yang khas adalah amaurosis kongenital Leber akibat mutasi CRX. Mutasi ini juga dapat muncul sebagai kasus sporadik ketika tidak ada anggota keluarga lain yang terkena.
Heterozigot majemuk: Pada penyakit autosomal resesif, bentuk ini terjadi ketika dua mutasi berbeda muncul pada dua alel. Seiring menurunnya perkawinan antarsepupu akhir-akhir ini, proporsi heterozigot majemuk meningkat dibandingkan homozigot.
Disomi uniparental: Fenomena ketika kedua salinan kromosom yang sama diwarisi dari satu orang tua, sementara kromosom dari orang tua lainnya tidak ada. Hal ini dapat muncul sebagai kasus yang tampaknya sporadik10).
Lebih dari 100 gen penyebab retinitis pigmentosa telah diidentifikasi, dan fenotipe yang sama pun dapat disebabkan oleh banyak gen yang berbeda2). Keragaman genetik ini membuat diagnosis menjadi sulit.
4. Praktik konseling genetik dan pemeriksaan genetik
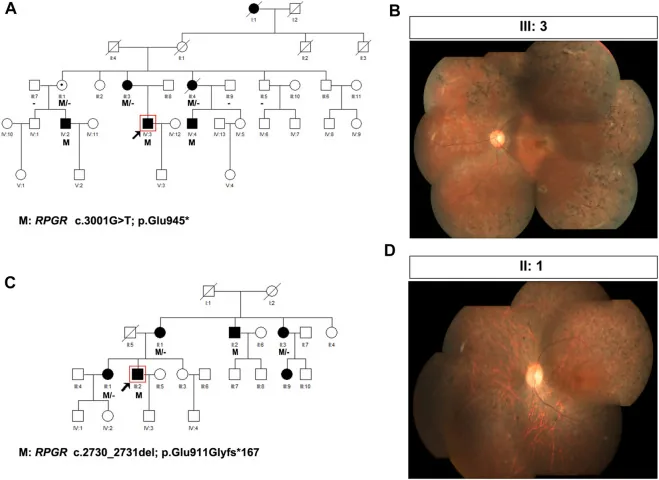
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Silsilah dan foto fundus dua keluarga dengan retinitis pigmentosa terkait X (varian RPGR: c.3001G>T dan c.2730_2731del). Silsilah menggunakan simbol standar (persegi terisi untuk laki-laki yang terkena, lingkaran bertitik untuk perempuan pembawa) untuk menunjukkan pola pewarisan, dan foto fundus yang sesuai menunjukkan pigmentasi spikula tulang yang lanjut dan atrofi retina. Ini sesuai dengan pembuatan pedigree dan penentuan pola pewarisan yang dibahas dalam bagian “4. Praktik konseling genetik dan pemeriksaan genetik”.
Untuk menjalankan konseling genetik dengan tepat, diperlukan persiapan berikut.
Membuat pedigree: Mengumpulkan riwayat keluarga setidaknya tiga generasi dan mencatatnya sebagai pedigree. Sebaran penderita secara vertikal dan horizontal dapat membantu memperkirakan pola pewarisan.
Memperkirakan pola pewarisan: Berdasarkan pedigree dan fenotipe, tentukan apakah bersifat autosomal dominan, autosomal resesif, terkait X, atau maternal.
Penjelasan tes genetik dan persetujuan tertulis: Sebelum memeriksa informasi genetik pasien, perlu memberikan penjelasan yang memadai tentang makna dan masalah dari pemeriksaan serta memperoleh persetujuan tertulis.
Anonimisasi hasil tes DNA dan pengelolaan informasi: Melindungi privasi melalui anonimisasi yang dapat ditautkan.
Menentukan terlebih dahulu kebijakan untuk temuan insidental: Mengonfirmasi dengan pasien sebelum pemeriksaan mengenai kebijakan pemberitahuan bila ditemukan temuan insidental serius yang dapat mengancam nyawa.
Akurat tinggi. Cocok untuk memastikan lokasi mutasi yang sudah diketahui
Pemeriksaan panel (targeted sequencing)
Kelompok gen yang terkait dengan penyakit tertentu
Pemeriksaan serentak gen yang terkait dengan penyakit retina. Tingkat diagnosis tinggi5)
Analisis eksom (NGS)
Seluruh wilayah ekson
Mendeteksi varian yang belum diketahui. Berguna jika ada banyak gen penyebab penyakit
Analisis seluruh genom (WGS)
Seluruh genom
Mungkin memiliki tingkat diagnosis lebih tinggi daripada pemeriksaan NGS bertarget yang ada13)
Pada distrofi retina herediter, ketika dicurigai penyakit akibat mutasi gen RPE65, dan dalam kasus seperti untuk membantu menentukan kelayakan terapi gen, beberapa pemeriksaan yang memenuhi syarat dapat dilakukan sebagai layanan medis yang ditanggung asuransi4).
Interpretasi varian memerlukan penilaian yang cermat. Diketahui bahwa sekitar 30% keterangan yang diterbitkan sebagai “varian” dalam artikel sebenarnya adalah polimorfisme (varian normal). Secara umum, perubahan urutan basa yang muncul pada 1 orang atau lebih dari setiap 100 orang sehat sebaiknya dianggap sebagai polimorfisme.
Basis data utama berikut digunakan.
OMIM (Online Mendelian Inheritance in Man): basis data komprehensif tentang penyakit dan gen herediter
GeneReviews: menyediakan informasi konseling genetik untuk tiap penyakit
RetNet (Retinal Information Network): basis data gen yang khusus untuk penyakit retina
QApakah biaya tes genetik ditanggung asuransi?
A
Pada beberapa penyakit mata herediter, tes genetik dapat dilakukan sebagai layanan medis yang ditanggung asuransi ketika indikasi terapi sedang ditentukan dan persyaratan fasilitas yang ditetapkan terpenuhi. Namun, apakah ditanggung atau tidak bergantung pada jenis tes dan penyakit yang diperiksa, sehingga perlu dikonfirmasi di fasilitas yang dikunjungi. Tes yang tidak ditanggung asuransi mungkin menjadi biaya pribadi. Pada penyakit tertentu yang ditetapkan sebagai penyakit sulit diobati (seperti retinitis pigmentosa), dukungan keuangan melalui sistem subsidi biaya medis untuk penyakit sulit diobati yang ditetapkan juga mungkin tersedia4).
5. Sistem konseling genetik dan prospek pengobatan
Dalam pelaksanaan konseling genetik, kolaborasi dengan dokter spesialis genetika dan konselor genetik tersertifikasi dianggap ideal. Di rumah sakit universitas, peninjauan oleh komite etik mungkin diperlukan. Karena informasi genetik dapat memengaruhi tidak hanya pasien tetapi juga anggota keluarga, diperlukan perhatian khusus dalam penanganannya. Diskusi sosial tentang pencegahan diskriminasi yang tidak adil berdasarkan informasi genetik juga terus berkembang8).
Penyakit langka yang ditetapkan dan bantuan biaya medis
Pada pasien dengan penyakit langka, beban biaya medis pribadi dikurangi. Ditetapkan batas bulanan untuk total biaya rawat jalan, rawat inap, dan obat, serta diterapkan kategori berdasarkan pendapatan.
Obat terapi gen voretigene neparvovec untuk distrofi retina herediter akibat mutasi gen RPE65 adalah sediaan yang diberikan secara subretina dengan vektor AAV2. Khasiat dan keamanannya telah dikonfirmasi dalam uji coba terkontrol acak6). Penentuan apakah terapi ini diindikasikan memerlukan penegasan tipe penyakit melalui pemeriksaan genetik.
Untuk terapi oligonukleotida antisense (ASO), pemberian intravitreal dari sediaan untuk Leber congenital amaurosis tipe 10 (LCA10) yang disebabkan oleh mutasi CEP290 sedang diteliti dalam uji klinis9).
Transplantasi sel epitel pigmen retina autologus menggunakan sel iPS sedang diteliti sebagai pengobatan regeneratif untuk degenerasi makula terkait usia7). Meskipun merupakan bidang yang berbeda dari penyakit retina herediter, hal ini menarik perhatian sebagai contoh nyata terapi sel retina.
QApakah terapi gen sudah tersedia sekarang?
A
Pada distrofi retina herediter akibat mutasi RPE65, efektivitas dan keamanan voretigene neparvovec telah ditunjukkan dalam RCT6). Untuk memastikan kelayakan terapi, diperlukan penentuan jenis penyakit melalui pemeriksaan genetik. Pada LCA10 dengan mutasi CEP290, injeksi intravitreal sediaan ASO sedang dipelajari dalam uji klinis9).
Pewarisan autosomal dominan, autosomal resesif, dan terpaut X mengikuti hukum Mendel. Namun, faktor-faktor berikut dapat membuat prediksi sederhana menjadi sulit.
Penetransi: meskipun memiliki mutasi, tidak semua orang akan mengalami penyakit. Bila penetransinya rendah, sifat dapat melompati generasi sehingga pola pewarisan sulit diperkirakan dari silsilah.
Ekspresivitas: bahkan di antara anggota keluarga dengan gen mutasi yang sama, tingkat keparahan gejala dapat berbeda.
Mutasi gain-of-function: mekanisme ketika protein yang bermutasi memperoleh fungsi baru yang merugikan. Ini berbeda dari efek dominan negatif yang biasa.
Mitokondria berada di sitoplasma, dan hanya mtDNA yang berasal dari sel telur ibu yang diwariskan kepada anak. Di setiap sel terdapat ribuan salinan mtDNA, dan keadaan ketika mtDNA mutan dan mtDNA normal bercampur (heteroplasmi) dapat terjadi. Semakin tinggi proporsi heteroplasmi, gejala cenderung semakin berat. Pada Leber hereditary optic neuropathy (LHON), tiga mutasi—11778 (paling sering), 3460, dan 14484—mencakup sekitar 90% dari seluruh mutasi.
Disomi uniparental adalah keadaan ketika sepasang kromosom berasal dari orang tua yang sama, tanpa kromosom dari orang tua lainnya. Anak yang lahir dari pembawa penyakit autosomal resesif dapat mengalami penyakit meskipun orang tua lainnya bukan pembawa, sehingga tampak sebagai kasus sporadis10). Bersama dengan mutasi de novo dan heterozigot majemuk, penyakit herediter perlu dipertimbangkan meskipun tidak ada riwayat keluarga.
Pada LCA10 dengan mutasi CEP290, koreksi splicing melalui terapi ASO sedang diteliti dalam uji klinis9).
Kandidat terapi penyuntingan genom EDIT-101 dengan CRISPR/Cas9 telah dilaporkan dalam pengembangan praklinis untuk LCA10 (mutasi CEP290)11).
Pengujian genetik praimplantasi (PGT-M) dapat dilakukan untuk penyakit herediter autosomal dominan dan resesif, dan dapat dipertimbangkan dalam kerangka etika12).
Whole-genome sequencing (WGS) telah ditunjukkan berpotensi meningkatkan tingkat diagnosis molekuler pada penyakit retina herediter dibandingkan dengan pemeriksaan genetik standar yang ada13).
Alat prediksi patogenisitas varian gen dengan memanfaatkan kecerdasan buatan (AI) sedang dikembangkan, dan diharapkan meningkatkan akurasi interpretasi varian.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.