Tipe tipikal (DOA)
Gejala: Penurunan visus bilateral progresif lambat.
Temuan papil: Pucat berbentuk baji temporal merupakan temuan sentral.
Gejala sistemik: Hanya atrofi saraf optik.
Visus: Lebih dari 80% mempertahankan visus ≥ 0,1.

Atrofi Optik Dominan Autosomal (Autosomal Dominant Optic Atrophy; ADOA) adalah neuropati optik herediter yang ditandai dengan atrofi optik progresif bilateral. Nomor OMIM adalah 165500. Ini adalah neuropati optik herediter yang paling sering, dengan perkiraan prevalensi 1:12.000 hingga 1:50.000 2).
Neuropati optik herediter secara luas dibagi menjadi ADOA (gen inti, pewarisan Mendel) dan Neuropati Optik Herediter Leber (LHON) (mutasi DNA mitokondria, pewarisan maternal). Atrofi optik resesif autosomal termasuk sindrom Wolfram. ADOA dan LHON sama-sama memiliki patologi degenerasi sel ganglion retina (RGC) akibat disfungsi mitokondria, tetapi gambaran klinis dan penyebab genetiknya berbeda 6).
Prevalensi diperkirakan 1:12.000 hingga 1:50.000, dan ini adalah yang paling sering di antara neuropati optik herediter 2). Prevalensi keseluruhan neuropati optik herediter di Inggris dilaporkan sekitar 1:25.000 6).
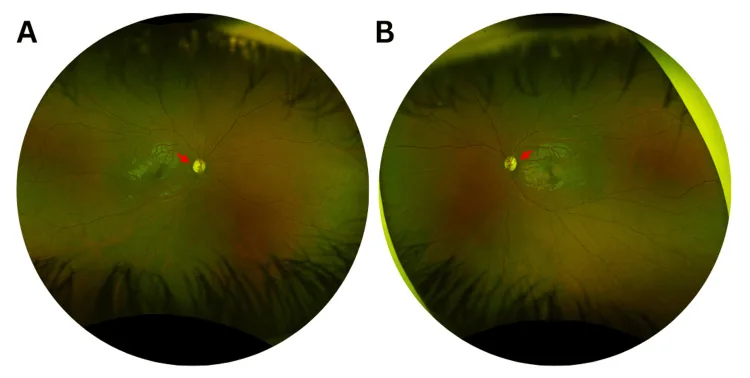
Onset tipikal terjadi pada dekade pertama hingga kedua kehidupan. Karena perjalanannya lambat, banyak pasien tidak dapat menentukan waktu onset secara tepat.
Ya. Dalam laporan Tachibana dkk. (2025), mutasi baru pada OPA1 diidentifikasi pada kasus yang mempertahankan ketajaman penglihatan terkoreksi terbaik 0,8/0,6 pada usia 56 tahun 2). Penetrasi berkisar antara 43–100%, dan kelainan hanya dapat dideteksi dengan OCT pada pembawa tanpa gejala.
Tiga temuan utama adalah papil saraf optik, penglihatan warna, dan lapang pandang.
ADOA memiliki fenotipe DOA plus yang disertai kelainan sistemik selain atrofi saraf optik. 20-30% pasien mengalami komplikasi seperti gangguan pendengaran, neuropati perifer, miopati, ataksia, dan oftalmoplegia eksternal progresif kronis (CPEO) 3).
Tipe tipikal (DOA)
Gejala: Penurunan visus bilateral progresif lambat.
Temuan papil: Pucat berbentuk baji temporal merupakan temuan sentral.
Gejala sistemik: Hanya atrofi saraf optik.
Visus: Lebih dari 80% mempertahankan visus ≥ 0,1.
DOA plus
Frekuensi: Muncul pada 20-30% pasien 3).
Komplikasi: Gangguan pendengaran, neuropati perifer, miopati, ataksia, CPEO, dll.
Tipe berat: Mutasi OPA1 bialelik menyebabkan sindrom Behr (onset dini, gangguan visus berat, ataksia, kejang) 3).
Lebih dari 60% kasus ADOA disebabkan oleh mutasi gen OPA1 6). OPA1 terletak pada kromosom 3q28-q29, dan lebih dari 500 mutasi patogenik telah diidentifikasi 6). Di Jepang, mutasi c.2708_2711 delTTAG dikenal sebagai mutasi frekuensi tinggi.
Pada kasus negatif OPA1, perlu dicari mutasi gen lain. Berikut adalah gen penyebab utama.
| Gen | Penyakit terkait | Catatan |
|---|---|---|
| OPA1 | ADOA (tipe tipikal / DOA plus) | Lebih dari 60% dari total 6) |
| AFG3L2 | Atrofi optik tipe 12 (OAT12), SCA28 | Sekitar 3% dari neuropati optik herediter 1) |
| OPA3 | Atrofi optik dominan dengan katarak / gangguan pendengaran (sindrom Costeff) | OMIM #258501 |
| WFS1 | Mirip sindrom Wolfram | OMIM #222370, #614296 |
| DNM1L | Atrofi saraf optik tipe 5 (OPA5) | Kontrol pembelahan mitokondria |
Brodsky dkk. (2023) mengidentifikasi atrofi saraf optik tipe 12 akibat mutasi c.1064C>T (p.Thr355Met) pada gen AFG3L2 pada seorang ayah dan anak perempuan keturunan Afrika Timur (Somalia)1). Dalam studi kohort 2186 kasus, AFG3L2 termasuk dalam 10 gen penyebab neuropati optik herediter teratas, mencakup 14 dari 451 kasus (3%).
Ya. Beberapa gen telah diidentifikasi seperti AFG3L2, OPA3, WFS1, dan DNM1L (OPA5). Khususnya, AFG3L2 mencakup sekitar 3% kasus neuropati optik herediter1), dan jika OPA1 negatif, pencarian komprehensif dengan sekuensing eksom/genom sangat penting.
Jika ditemukan gangguan penglihatan bilateral yang tidak dapat dijelaskan pada usia sekolah, curigai penyakit ini. Riwayat keluarga dengan gejala serupa adalah petunjuk penting, tetapi mungkin tidak ada riwayat keluarga karena penetrasi tidak lengkap.
Pada kasus pria 56 tahun oleh Tachibana dkk. (2025), lapang pandang HFA 24-2 stimulus putih normal, tetapi lapang pandang blue-on-yellow mendeteksi penurunan sensitivitas2). OCT menunjukkan penipisan RNFL temporal, CFF menurun menjadi 30/31 Hz (normal >39 Hz).
| Penyakit | Pola onset | Pola pewarisan | Poin pembeda |
|---|---|---|---|
| LHON | Akut hingga subakut | Warisan ibu | Predileksi pria muda, berat |
| ADOA | Lambat dan tersembunyi | Autosomal dominan | Masa kanak-kanak, bilateral simetris |
| Glaukoma | Lambat | Multifaktorial | Peningkatan tekanan intraokular, perluasan cekungan papil |
| Neuropati optik kompresif | Lambat hingga subakut | Non-herediter | Lesi kiasma optikum pada MRI |
Diagnosis banding lainnya: neuropati optik toksik (misalnya etambutol), neuropati optik defisiensi nutrisi, distrofi makula okult, distrofi kerucut, ambliopia fungsional, gangguan penglihatan psikogenik.
Tidak ada terapi efektif yang terbukti. Perawatan penglihatan rendah dan konseling pasien menjadi andalan terapi.
Untuk mengurangi stres oksidatif, berikut telah diusulkan, namun belum ada yang ditetapkan sebagai terapi standar.
Penting untuk menjelaskan pewarisan dominan autosomal dan memberikan informasi tentang risiko keparahan akibat mutasi bialelik (sindrom Behr) 3).
Penurunan penglihatan umumnya berlangsung lambat dan lebih ringan dibandingkan LHON. Lebih dari 80% pasien mempertahankan ketajaman penglihatan terkoreksi 0,1 atau lebih, namun ada kasus yang turun menjadi 0,1 atau kurang. Karena onset lambat, kesadaran mungkin tertunda, dan kadang ditemukan secara tidak sengaja saat pemeriksaan. Karena tidak ada terapi yang terbukti, pemantauan rutin dan perawatan low vision jangka panjang penting.
Saat ini belum ada terapi efektif yang mapan. Perawatan low vision menjadi andalan pengobatan. Idebenone dilaporkan berpotensi menstabilkan atau memulihkan penglihatan 4)5), namun belum ditetapkan sebagai terapi standar. Untuk pendekatan terapi yang masih dalam tahap penelitian, lihat bagian “Penelitian Terkini dan Prospek Masa Depan”.
OPA1 adalah GTPase terkait dinamin yang berada di membran dalam mitokondria. Disintesis di inti sel kemudian diangkut ke mitokondria, dan menjalankan fungsi berikut 6).
Mekanisme patologis utama adalah haploinsufisiensi. Sebagian besar mutasi OPA1 menyebabkan terminasi dini translasi, sehingga jumlah protein OPA1 berkurang 6).
Penurunan protein OPA1 → Peningkatan fragmentasi mitokondria dan peningkatan daur ulang 3) → Gangguan metabolisme mitokondria dan gangguan fosforilasi oksidatif → Peningkatan spesies oksigen reaktif (ROS) → Apoptosis sel ganglion retina (RGC).
Terutama sel ganglion retina di berkas papilomakular mengalami degenerasi, yang tampak sebagai atrofi saraf optik temporal.
AFG3L2 mengkode subunit dari protease AAA metalloprotease matriks mitokondria (m-AAA) 1). Membentuk kompleks dengan SPG7 (paraplegin) dan melakukan pemrosesan, pematangan, dan kontrol kualitas protein mitokondria secara ATP-dependen. Mutasi menyebabkan degenerasi RGC serupa dengan OPA1 1).
Terapi ASO
Target: Ekson penginduksi NMD (degradasi yang bergantung pada nonsense) dari pre-mRNA OPA1.
Mekanisme: Menghambat penyertaan ekson yang menginduksi NMD, sehingga meningkatkan translasi OPA1 tipe liar melalui pendekatan yang tidak bergantung pada mutasi 6).
Status Saat Ini: Peningkatan produksi protein OPA1 dan perbaikan bioenergetika mitokondria telah dikonfirmasi pada tiga galur sel yang berasal dari pasien ADOA 6). Memerlukan injeksi intravitreal berulang secara teratur, dengan risiko endoftalmitis dan uveitis kronis sebagai tantangan.
Terapi Gen
Model Tikus: Terapi gen OPA1 mencegah kehilangan sel ganglion retina pada model tikus DOA 7).
Optimalisasi Isoform: Isoform OPA1 yang dioptimalkan 1 dan 7 menunjukkan efek terapeutik pada model disfungsi mitokondria 8).
Status Saat Ini: Tahap praklinis.
Penyuntingan Gen
CRISPR-Cas9: Koreksi mutasi OPA1 c.1334G>A: p.R445H pada iPSC memulihkan homeostasis mitokondria 9).
Trans-splicing: Pendekatan koreksi mutasi patogenik pada tingkat mRNA juga sedang diteliti 6).
Sel punca: Regenerasi saraf optik menggunakan RGC yang berasal dari iPSC sedang diteliti dalam studi praklinis 6).
Analisis ulang sekuensing eksom secara berulang juga merupakan kemajuan diagnostik yang penting. Seiring meluasnya pengetahuan genetik, diagnosis baru dapat diperoleh dari data yang sebelumnya negatif pada pemeriksaan sebelumnya 1).