النمط النموذجي (DOA)
الأعراض: تدهور بصري تدريجي بطيء في كلتا العينين.
علامات القرص البصري: الشحوب الإسفيني الصدغي هو العلامة المركزية.
الأعراض الجهازية: ضمور العصب البصري فقط.
حدة البصر: أكثر من 80% يحافظون على حدة بصر ≥ 0.1.

الضمور البصري السائد الجسدي (Autosomal Dominant Optic Atrophy; ADOA) هو اعتلال عصبي بصري وراثي يتميز بضمور بصري تقدمي ثنائي. رقم OMIM هو 165500. وهو أكثر الاعتلالات العصبية البصرية الوراثية شيوعًا، ويقدر معدل الانتشار بـ 1:12,000 إلى 1:50,000 2).
تنقسم الاعتلالات العصبية البصرية الوراثية بشكل كبير إلى ADOA (الجينات النووية، وراثة مندلية) واعتلال ليبر العصبي البصري الوراثي (LHON) (طفرات الحمض النووي للميتوكوندريا، وراثة أمومية). يشمل ضمور العصب البصري المتنحي الجسدي متلازمة ولفرام. يشترك كل من ADOA وLHON في نفس الآلية المرضية لتنكس الخلايا العقدية الشبكية (RGC) بسبب خلل وظيفي في الميتوكوندريا، لكن الصور السريرية والأسباب الوراثية مختلفة 6).
يقدر معدل الانتشار بـ 1:12,000 إلى 1:50,000، وهو الأكثر شيوعًا بين الاعتلالات العصبية البصرية الوراثية 2). تم الإبلاغ عن معدل انتشار إجمالي للاعتلالات العصبية البصرية الوراثية في المملكة المتحدة بحوالي 1:25,000 6).
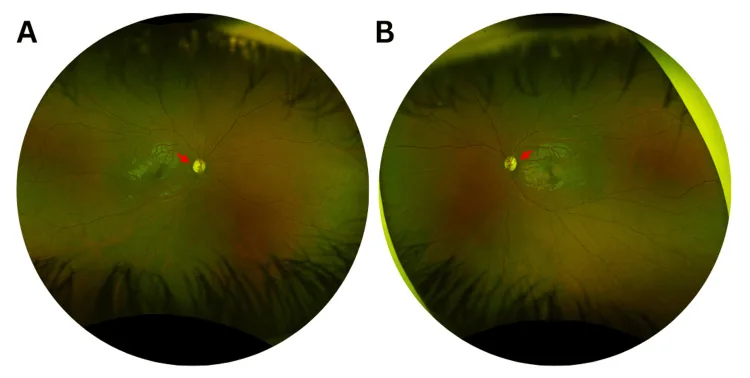
تبدأ الحالة عادةً في العقد الأول أو الثاني من العمر. نظرًا للتقدم البطيء، لا يستطيع العديد من المرضى تحديد وقت البداية بدقة.
نعم. في تقرير Tachibana وآخرين (2025)، تم تحديد طفرة جديدة في OPA1 في حالة حافظت على أفضل حدة رؤية مصححة 0.8/0.6 في سن 56 2). تتراوح النفاذية بين 43% و100%، وقد يتم اكتشاف تشوهات فقط بواسطة OCT في الناقلين بدون أعراض.
النتائج الرئيسية الثلاثة هي قرص العصب البصري ورؤية الألوان ومجال الرؤية.
يوجد لـ ADOA نمط ظاهري DOA plus يرافقه تشوهات جهازية غير ضمور العصب البصري. 20-30% من المرضى يعانون من مضاعفات مثل فقدان السمع، الاعتلال العصبي المحيطي، الاعتلال العضلي، الرنح، وشلل العين الخارجي المزمن التدريجي (CPEO) 3).
النمط النموذجي (DOA)
الأعراض: تدهور بصري تدريجي بطيء في كلتا العينين.
علامات القرص البصري: الشحوب الإسفيني الصدغي هو العلامة المركزية.
الأعراض الجهازية: ضمور العصب البصري فقط.
حدة البصر: أكثر من 80% يحافظون على حدة بصر ≥ 0.1.
DOA بلس
التكرار: يظهر في 20-30% من المرضى 3).
المضاعفات: فقدان السمع، الاعتلال العصبي المحيطي، الاعتلال العضلي، الرنح، CPEO، وغيرها.
النمط الشديد: تؤدي طفرات OPA1 ثنائية الأليل إلى متلازمة بيهر (بداية مبكرة، ضعف بصري شديد، رنح، تشنجات) 3).
أكثر من 60% من حالات ADOA ناتجة عن طفرات في جين OPA1 6). يقع OPA1 على الكروموسوم 3q28-q29، وتم تحديد أكثر من 500 طفرة ممرضة 6). في اليابان، تُعرف طفرة c.2708_2711 delTTAG كطفرة عالية التكرار.
في الحالات السلبية لـ OPA1، يجب البحث عن طفرات جينية أخرى. فيما يلي الجينات المسببة الرئيسية.
| الجين | المرض المرتبط | ملاحظات |
|---|---|---|
| OPA1 | ADOA (النمط النموذجي / DOA بلس) | أكثر من 60% من الحالات 6) |
| AFG3L2 | ضمور العصب البصري من النوع 12 (OAT12)، SCA28 | حوالي 3% من حالات الاعتلال العصبي البصري الوراثي 1) |
| OPA3 | ضمور العصب البصري السائد مع إعتام عدسة العين / فقدان السمع (متلازمة كوستيف) | OMIM #258501 |
| WFS1 | متلازمة ولفرام الشبيهة | OMIM #222370, #614296 |
| DNM1L | ضمور العصب البصري من النوع 5 (OPA5) | التحكم في انقسام الميتوكوندريا |
حدد Brodsky وآخرون (2023) ضمور العصب البصري من النوع 12 الناجم عن طفرة c.1064C>T (p.Thr355Met) في جين AFG3L2 لدى أب وابنته من أصل شرق أفريقي (صومالي)1). في دراسة جماعية شملت 2186 حالة، كان AFG3L2 ضمن أفضل 10 جينات مسببة لاعتلال العصب البصري الوراثي، وشكل 14 من 451 حالة (3%).
نعم. تم تحديد عدة جينات مثل AFG3L2 وOPA3 وWFS1 وDNM1L (OPA5). على وجه الخصوص، يشكل AFG3L2 حوالي 3% من حالات اعتلال العصب البصري الوراثي1)، وعندما تكون نتيجة OPA1 سلبية، يكون البحث الشامل باستخدام تسلسل الإكسوم أو الجينوم مهمًا.
إذا تم اكتشاف ضعف بصري ثنائي غير مفسر في سن المدرسة، يجب الاشتباه في هذا المرض. التاريخ العائلي لأعراض مماثلة هو دليل مهم، ولكن قد لا يكون هناك تاريخ عائلي بسبب ضعف النفاذية.
في حالة رجل يبلغ من العمر 56 عامًا من Tachibana وآخرين (2025)، كان مجال الرؤية HFA 24-2 بالتحفيز الأبيض طبيعيًا، لكن المجال البصري الأزرق على الأصفر كشف عن انخفاض الحساسية2). أظهر OCT ترقق RNFL الصدغي، وانخفاض CFF إلى 30/31 هرتز (الطبيعي >39 هرتز).
| المرض | نمط البداية | نمط الوراثة | نقاط التمايز |
|---|---|---|---|
| LHON | حاد إلى تحت حاد | وراثة أمومية | شائع لدى الذكور الشباب، شديد |
| ADOA | بطيء وكامن | جسمي سائد | الطفولة، ثنائي ومتناظر |
| الزرق | بطيء | متعدد العوامل | ارتفاع ضغط العين، توسع حفرة العصب البصري |
| اعتلال العصب البصري الانضغاطي | بطيء إلى تحت الحاد | غير وراثي | آفة التصالب البصري في التصوير بالرنين المغناطيسي |
تشخيصات تفريقية أخرى: الاعتلال العصبي البصري السام (مثل إيثامبوتول)، الاعتلال العصبي البصري الناتج عن نقص التغذية، الحثل البقعي الخفي، الحثل المخروطي، الغمش الوظيفي، الاضطراب البصري النفسي.
لا يوجد علاج فعال مثبت. الرعاية البصرية المنخفضة واستشارة المرضى هما أساس العلاج.
لتقليل الإجهاد التأكسدي، تم اقتراح ما يلي، لكن لم يتم إثبات أي منها كعلاج قياسي.
من المهم شرح أن الوراثة هي صبغية جسدية سائدة، وتقديم معلومات عن خطر الشدة بسبب الطفرات ثنائية الأليل (متلازمة بهر) 3).
يتطور فقدان البصر عادة ببطء، ويكون مساره أكثر اعتدالًا مقارنة باعتلال العصب البصري الوراثي ليبر (LHON). يحافظ أكثر من 80% من المرضى على حدة بصر مصححة تبلغ 0.1 أو أفضل، لكن هناك حالات تنخفض فيها حدة البصر المصححة إلى 0.1 أو أقل. نظرًا لأن البداية بطيئة، قد يتأخر الوعي، ويتم اكتشافه أحيانًا بالصدفة أثناء الفحوصات. نظرًا لعدم وجود علاج مثبت، فإن المراقبة المنتظمة ورعاية ضعف البصر على المدى الطويل مهمة.
لا يوجد علاج فعال مثبت حتى الآن. الرعاية البصرية المنخفضة هي أساس العلاج. هناك تقارير تشير إلى أن الإيديبينون قد يحقق استقرارًا أو تحسنًا في الرؤية 4)5)، لكنه لم يثبت كعلاج قياسي. بالنسبة للنهج العلاجية قيد البحث، انظر قسم “أحدث الأبحاث والآفاق المستقبلية”.
OPA1 هو بروتين GTPase مرتبط بالدينامين موجود في الغشاء الداخلي للميتوكوندريا. يتم تصنيعه في النواة ثم نقله إلى الميتوكوندريا، حيث يؤدي الوظائف التالية 6).
الآلية المرضية الرئيسية هي نقص الكفاءة (haploinsufficiency). معظم طفرات OPA1 تؤدي إلى إنهاء مبكر للترجمة، مما يسبب نقصًا في كمية بروتين OPA1 6).
انخفاض بروتين OPA1 → زيادة تجزؤ الميتوكوندريا وزيادة إعادة التدوير 3) → اضطراب أيض الميتوكوندريا وخلل في الفسفرة التأكسدية → ارتفاع أنواع الأكسجين التفاعلية (ROS) → موت الخلايا المبرمج للخلايا العقدية الشبكية (RGC).
تتحلل الخلايا العقدية الشبكية في الحزمة الحليمية البقعية بشكل رئيسي، مما يظهر كضمور العصب البصري الصدغي.
يشفر AFG3L2 الوحدة الفرعية لبروتياز الميتوكوندريا من نوع AAA (m-AAA) في المطرس 1). يشكل معقدًا مع SPG7 (paraplegin) ويقوم بمعالجة ونضج ومراقبة جودة بروتينات الميتوكوندريا بطريقة تعتمد على ATP. تؤدي الطفرات إلى تنكس الخلايا العقدية الشبكية مثل OPA1 1).
علاج ASO
الهدف: إكسون تحفيز تحلل الـ NMD (التحلل المعتمد على الهراء) لـ pre-mRNA الخاص بـ OPA1.
الآلية: تثبيط إدراج الإكسون الذي يحفز NMD، مما يعزز ترجمة OPA1 من النوع البري بطريقة غير معتمدة على الطفرة 6).
الوضع الحالي: تم تأكيد زيادة إنتاج بروتين OPA1 وتحسين الطاقة الحيوية للميتوكوندريا في ثلاث سلالات خلايا مشتقة من مرضى ADOA 6). يتطلب الحقن الزجاجي المتكرر، مع مخاطر التهاب باطن العين والتهاب العنبية المزمن كتحديات.
العلاج الجيني
نموذج الفأر: العلاج الجيني لـ OPA1 يمنع فقدان الخلايا العقدية الشبكية في نموذج فأر DOA 7).
تحسين الأشكال الإسوية: الأشكال الإسوية المحسنة 1 و7 من OPA1 تظهر تأثيرًا علاجيًا في نماذج خلل وظيفة الميتوكوندريا 8).
الوضع الحالي: مرحلة ما قبل السريرية.
تحرير الجينات
CRISPR-Cas9: تصحيح طفرة OPA1 c.1334G>A: p.R445H في الخلايا الجذعية المحفزة يستعيد توازن الميتوكوندريا 9).
الربط العابر: يتم أيضًا دراسة نهج تصحيح الطفرات المسببة للأمراض على مستوى mRNA 6).
الخلايا الجذعية: يتم دراسة تجديد العصب البصري باستخدام خلايا RGC المشتقة من iPSC في الدراسات قبل السريرية 6).
إعادة التحليل المتكرر لتسلسل الإكسوم هو أيضًا تقدم تشخيصي مهم. مع توسع المعرفة الوراثية، قد يتم الحصول على تشخيصات جديدة من بيانات كانت سلبية في الفحوصات السابقة 1).