ชนิดทั่วไป (DOA)
อาการ: การมองเห็นลดลงทั้งสองข้างแบบค่อยเป็นค่อยไป
ลักษณะของจานประสาทตา: สีซีดรูปลิ่มทางขมับเป็นลักษณะสำคัญ
อาการทั่วร่างกาย: มีเพียงฝ่อของเส้นประสาทตา
ความคมชัดของการมองเห็น: มากกว่า 80% รักษาระดับการมองเห็น ≥ 0.1

โรคจอประสาทตาฝ่อแบบ autosomal dominant (Autosomal Dominant Optic Atrophy; ADOA) เป็นโรคเส้นประสาทตาทางพันธุกรรมที่มีลักษณะเฉพาะคือ จอประสาทตาฝ่อแบบลุกลามทั้งสองข้าง หมายเลข OMIM คือ 165500 เป็นโรคเส้นประสาทตาทางพันธุกรรมที่พบบ่อยที่สุด โดยมีความชุกประมาณ 1:12,000 ถึง 1:50,000 2)
โรคเส้นประสาทตาทางพันธุกรรมแบ่งออกเป็น ADOA (ยีนในนิวเคลียส การถ่ายทอดแบบเมนเดเลียน) และโรคเส้นประสาทตาเสื่อมทางพันธุกรรมของ Leber (LHON) (การกลายพันธุ์ของ DNA ในไมโตคอนเดรีย การถ่ายทอดทางมารดา) จอประสาทตาฝ่อแบบ autosomal recessive ได้แก่ กลุ่มอาการ Wolfram ทั้ง ADOA และ LHON มีพยาธิสภาพร่วมกันคือการเสื่อมของเซลล์ปมประสาทจอตา (RGC) เนื่องจากความผิดปกติของไมโตคอนเดรีย แต่ลักษณะทางคลินิกและสาเหตุทางพันธุกรรมแตกต่างกัน 6)
ความชุกประมาณ 1:12,000 ถึง 1:50,000 และเป็นโรคเส้นประสาทตาทางพันธุกรรมที่พบบ่อยที่สุด 2) มีรายงานความชุกโดยรวมของโรคเส้นประสาทตาทางพันธุกรรมในสหราชอาณาจักรประมาณ 1:25,000 6)
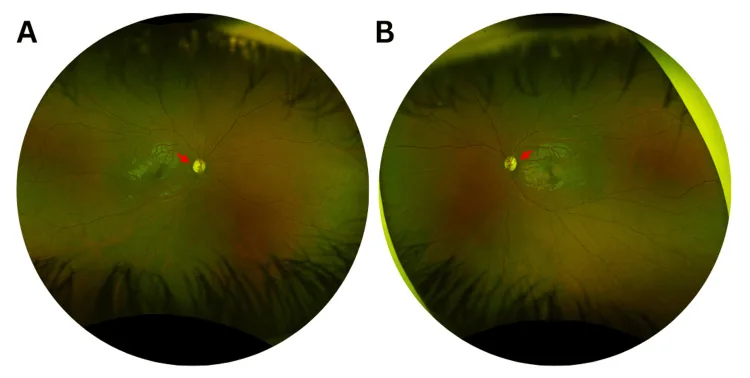
การเริ่มต้นโดยทั่วไปเกิดขึ้นในทศวรรษแรกถึงทศวรรษที่สองของชีวิต เนื่องจากดำเนินไปอย่างช้าๆ ผู้ป่วยจำนวนมากจึงไม่สามารถระบุเวลาที่เริ่มต้นได้อย่างแม่นยำ
ได้ ในรายงานของ Tachibana และคณะ (2025) พบการกลายพันธุ์ใหม่ในยีน OPA1 ในผู้ป่วยที่รักษาค่าสายตาที่แก้ไขดีที่สุด 0.8/0.6 เมื่ออายุ 56 ปี 2) อัตราการแสดงออกของยีนอยู่ระหว่าง 43–100% และอาจตรวจพบความผิดปกติได้ด้วย OCT เท่านั้นในพาหะที่ไม่มีอาการ
อาการแสดงหลักสามประการคือ จานประสาทตา การมองเห็นสี และ ลานสายตา
ADOA มีฟีโนไทป์ DOA plus ซึ่งมีความผิดปกติทั่วร่างกายนอกเหนือจากฝ่อของเส้นประสาทตา ผู้ป่วย 20-30% มีภาวะแทรกซ้อน เช่น การสูญเสียการได้ยิน โรคเส้นประสาทส่วนปลาย โรคกล้ามเนื้อ ภาวะเสียการทรงตัว และกล้ามเนื้อตาภายนอกอ่อนแรงเรื้อรังแบบก้าวหน้า (CPEO) 3)
ชนิดทั่วไป (DOA)
อาการ: การมองเห็นลดลงทั้งสองข้างแบบค่อยเป็นค่อยไป
ลักษณะของจานประสาทตา: สีซีดรูปลิ่มทางขมับเป็นลักษณะสำคัญ
อาการทั่วร่างกาย: มีเพียงฝ่อของเส้นประสาทตา
ความคมชัดของการมองเห็น: มากกว่า 80% รักษาระดับการมองเห็น ≥ 0.1
DOA plus
ความถี่: เกิดขึ้นใน 20-30% ของผู้ป่วย 3)
ภาวะแทรกซ้อน: การสูญเสียการได้ยิน โรคเส้นประสาทส่วนปลาย โรคกล้ามเนื้อ ภาวะเสียการทรงตัว CPEO เป็นต้น
ชนิดรุนแรง: การกลายพันธุ์ OPA1 แบบสองอัลลีลทำให้เกิดกลุ่มอาการเบห์ร (เริ่มต้นเร็ว การมองเห็นบกพร่องรุนแรง ภาวะเสียการทรงตัว ชัก) 3)
มากกว่า 60% ของผู้ป่วย ADOA เกิดจากการกลายพันธุ์ของยีน OPA1 6) ยีน OPA1 อยู่บนโครโมโซม 3q28-q29 และมีการระบุการกลายพันธุ์ที่ก่อโรคมากกว่า 500 ชนิด 6) ในญี่ปุ่น การกลายพันธุ์ c.2708_2711 delTTAG เป็นที่รู้จักว่าเป็นการกลายพันธุ์ความถี่สูง
ในกรณีที่ผล OPA1 เป็นลบ จำเป็นต้องค้นหาการกลายพันธุ์ของยีนอื่นๆ ด้านล่างนี้คือยีนก่อโรคหลัก
| ยีน | โรคที่เกี่ยวข้อง | หมายเหตุ |
|---|---|---|
| OPA1 | ADOA (ชนิดทั่วไป / DOA plus) | มากกว่า 60% ของทั้งหมด 6) |
| AFG3L2 | จอประสาทตาฝ่อชนิดที่ 12 (OAT12), SCA28 | ประมาณ 3% ของโรคเส้นประสาทตาทางพันธุกรรม 1) |
| OPA3 | จอประสาทตาฝ่อแบบเด่นร่วมกับต้อกระจก / สูญเสียการได้ยิน (Costeff syndrome) | OMIM #258501 |
| WFS1 | คล้าย Wolfram syndrome | OMIM #222370, #614296 |
| DNM1L | จอประสาทตาฝ่อชนิดที่ 5 (OPA5) | การควบคุมการแบ่งตัวของไมโตคอนเดรีย |
Brodsky และคณะ (2023) ระบุจอประสาทตาฝ่อชนิดที่ 12 จากการกลายพันธุ์ c.1064C>T (p.Thr355Met) ในยีน AFG3L2 ในพ่อและลูกสาวเชื้อสายแอฟริกาตะวันออก (โซมาลี)1) ในการศึกษา cohort จำนวน 2186 ราย พบว่า AFG3L2 อยู่ใน 10 อันดับแรกของยีนที่ทำให้เกิดโรคเส้นประสาทตาทางพันธุกรรม คิดเป็น 14 จาก 451 ราย (3%)
มี มียีนหลายตัวที่ถูกระบุ เช่น AFG3L2, OPA3, WFS1 และ DNM1L (OPA5) โดยเฉพาะ AFG3L2 คิดเป็นประมาณ 3% ของโรคเส้นประสาทตาทางพันธุกรรม1) และเมื่อ OPA1 เป็นลบ การค้นหาอย่างครอบคลุมด้วยการหาลำดับเอ็กโซม/จีโนมเป็นสิ่งสำคัญ
หากพบความบกพร่องทางการมองเห็นทั้งสองข้างโดยไม่ทราบสาเหตุในวัยเรียน ควรสงสัยโรคนี้ ประวัติครอบครัวที่มีอาการคล้ายกันเป็นเบาะแสสำคัญ แต่อาจไม่มีประวัติครอบครัวเนื่องจาก incomplete penetrance
ในกรณีผู้ป่วยชายอายุ 56 ปีของ Tachibana และคณะ (2025) ลานสายตา HFA 24-2 ด้วยสิ่งเร้าสีขาวปกติ แต่ลานสายตาแบบ blue-on-yellow ตรวจพบการลดลงของความไว2) OCT แสดงการบางลงของ RNFL ขมับ CFF ลดลงเหลือ 30/31 Hz (ค่าปกติ >39 Hz)
| โรค | รูปแบบการเริ่มต้น | รูปแบบการถ่ายทอดทางพันธุกรรม | จุดที่ใช้แยก |
|---|---|---|---|
| LHON | เฉียบพลันถึงกึ่งเฉียบพลัน | ถ่ายทอดทางมารดา | พบบ่อยในชายหนุ่ม รุนแรง |
| ADOA | ช้าและแอบแฝง | ถ่ายทอดทาง autosomal dominant | วัยเด็ก, ตาทั้งสองข้างสมมาตร |
| ต้อหิน | ช้า | หลายปัจจัย | ความดันลูกตาสูง, การขยายของแอ่งประสาทตา |
| โรคเส้นประสาทตาถูกกดทับ | ช้าถึงกึ่งเฉียบพลัน | ไม่ใช่พันธุกรรม | รอยโรคที่ออปติกไคแอสมาบน MRI |
การวินิจฉัยแยกโรคอื่นๆ: โรคเส้นประสาทตาจากสารพิษ (เช่น ethambutol), โรคเส้นประสาทตาจากการขาดสารอาหาร, จอประสาทตาเสื่อมแบบซ่อนเร้น, จอประสาทตาเสื่อมแบบ cone dystrophy, ตาขี้เกียจจากการทำงาน, ความผิดปกติทางการมองเห็นจากจิตใจ
ไม่มีการรักษาที่มีประสิทธิภาพที่ได้รับการยืนยัน การดูแลสายตาเลือนรางและการให้คำปรึกษาผู้ป่วยเป็นหลักในการรักษา
เพื่อลดความเครียดออกซิเดชัน ได้มีการเสนอสิ่งต่อไปนี้ แต่ยังไม่มีสิ่งใดที่ถูกกำหนดให้เป็นการรักษามาตรฐาน
สิ่งสำคัญคือต้องอธิบายเกี่ยวกับการถ่ายทอดทางพันธุกรรมแบบออโตโซมอลโดมิแนนท์ และให้ข้อมูลเกี่ยวกับความเสี่ยงของความรุนแรงจากการกลายพันธุ์แบบสองอัลลีล (Behr syndrome) 3)
การสูญเสียการมองเห็นโดยทั่วไปจะดำเนินไปอย่างช้าๆ และมีแนวทางที่ไม่รุนแรงเมื่อเทียบกับ LHON ผู้ป่วยมากกว่า 80% ยังคงมีสายตาที่แก้ไขแล้วที่ 0.1 หรือดีกว่า แต่ก็มีบางกรณีที่ลดลงเหลือ 0.1 หรือน้อยกว่า เนื่องจากการเริ่มต้นช้า การรับรู้อาจล่าช้า และบางครั้งพบโดยบังเอิญระหว่างการตรวจ เนื่องจากไม่มีการรักษาที่เป็นมาตรฐาน การติดตามอย่างสม่ำเสมอและการดูแลผู้มีสายตาเลือนรางในระยะยาวจึงมีความสำคัญ
ปัจจุบันยังไม่มีการรักษาที่มีประสิทธิภาพที่ได้รับการยอมรับ การดูแลผู้ที่มีสายตาเลือนรางเป็นหลักในการรักษา มีรายงานว่าไอดีบีโนน (Idebenone) อาจช่วยให้การมองเห็นคงที่หรือฟื้นตัวได้ 4)5) แต่ยังไม่ได้รับการยอมรับให้เป็นการรักษามาตรฐาน สำหรับแนวทางการรักษาที่อยู่ในขั้นตอนการวิจัย โปรดดูหัวข้อ “งานวิจัยล่าสุดและแนวโน้มในอนาคต”
OPA1 เป็น GTPase ที่เกี่ยวข้องกับไดนามินซึ่งอยู่ที่เยื่อหุ้มชั้นในของไมโทคอนเดรีย ถูกสังเคราะห์ในนิวเคลียสแล้วส่งไปยังไมโทคอนเดรีย ทำหน้าที่ดังต่อไปนี้ 6)
กลไกทางพยาธิวิทยาหลักคือภาวะแฮพลอยด์ไม่เพียงพอ (haploinsufficiency) การกลายพันธุ์ของ OPA1 ส่วนใหญ่ทำให้เกิดการสิ้นสุดการแปลรหัสก่อนกำหนด ส่งผลให้ปริมาณโปรตีน OPA1 ลดลง 6)
การลดลงของโปรตีน OPA1 → การเพิ่มขึ้นของการแตกตัวของไมโทคอนเดรียและการหมุนเวียนที่เพิ่มขึ้น 3) → ความผิดปกติของเมแทบอลิซึมของไมโทคอนเดรียและการบกพร่องของออกซิเดทีฟฟอสโฟรีเลชัน → การเพิ่มขึ้นของอนุมูลอิสระ (ROS) → การตายแบบอะพอพโทซิสของเซลล์ปมประสาทจอตา (RGC)
เซลล์ปมประสาทจอตาในมัดประสาทตา-จุดรับภาพ (papillomacular bundle) เสื่อมเป็นหลัก ทำให้เกิดการฝ่อของเส้นประสาทตาด้านขมับ
AFG3L2 เข้ารหัสหน่วยย่อยของเมทัลโลโปรตีเอส AAA ในเมทริกซ์ไมโทคอนเดรีย (m-AAA) 1) สร้างสารเชิงซ้อนกับ SPG7 (paraplegin) และทำหน้าที่แปรรูป ทำให้สมบูรณ์ และควบคุมคุณภาพของโปรตีนไมโทคอนเดรียโดยอาศัย ATP การกลายพันธุ์ทำให้เกิดการเสื่อมของ RGC เช่นเดียวกับ OPA1 1)
การบำบัดด้วย ASO
เป้าหมาย: เอ็กซอนที่เหนี่ยวนำ NMD (การย่อยสลายที่ขึ้นกับ nonsense) ของ pre-mRNA OPA1
กลไก: ยับยั้งการรวมตัวของเอ็กซอนที่เหนี่ยวนำ NMD เพิ่มการแปลรหัส OPA1 ชนิดปกติโดยวิธีที่ไม่ขึ้นกับการกลายพันธุ์ 6)
สถานะปัจจุบัน: ยืนยันการเพิ่มการผลิตโปรตีน OPA1 และการปรับปรุงพลังงานชีวภาพของไมโตคอนเดรียในเซลล์สามสายพันธุ์ที่ได้จากผู้ป่วย ADOA 6) ต้องฉีดเข้าจอประสาทตาเป็นระยะ ความเสี่ยงของเยื่อบุตาอักเสบและม่านตาอักเสบเรื้อรังเป็นความท้าทาย
การบำบัดด้วยยีน
แบบจำลองหนู: การบำบัดด้วยยีน OPA1 ป้องกันการสูญเสียเซลล์ปมประสาทจอตาในแบบจำลองหนู DOA 7)
การปรับไอโซฟอร์มให้เหมาะสม: ไอโซฟอร์ม OPA1 ที่ปรับปรุงแล้ว 1 และ 7 แสดงผลการรักษาในแบบจำลองความผิดปกติของไมโตคอนเดรีย 8)
สถานะปัจจุบัน: ระยะก่อนคลินิก
การตัดต่อยีน
CRISPR-Cas9: การแก้ไขการกลายพันธุ์ OPA1 c.1334G>A: p.R445H ใน iPSC ช่วยฟื้นฟูสมดุลของไมโตคอนเดรีย 9)
Trans-splicing: วิธีการแก้ไขการกลายพันธุ์ที่ก่อโรคในระดับ mRNA กำลังอยู่ระหว่างการศึกษา 6)
สเต็มเซลล์: การฟื้นฟูเส้นประสาทตาด้วย RGC ที่ได้จาก iPSC กำลังอยู่ระหว่างการศึกษาในงานวิจัยก่อนทางคลินิก 6)
การวิเคราะห์ซ้ำของเอ็กโซมซีเควนซิงอย่างต่อเนื่องก็เป็นความก้าวหน้าทางการวินิจฉัยที่สำคัญเช่นกัน ด้วยการขยายความรู้ทางพันธุกรรม การวินิจฉัยใหม่อาจได้จากข้อมูลที่เคยให้ผลลบในการตรวจก่อนหน้านี้ 1)