Thể điển hình (DOA)
Triệu chứng: Giảm thị lực hai bên tiến triển chậm.
Dấu hiệu gai thị: Nhợt hình nêm thái dương là dấu hiệu trung tâm.
Triệu chứng toàn thân: Chỉ teo thần kinh thị.
Thị lực: Hơn 80% duy trì thị lực ≥ 0,1.

Bệnh teo thị thần kinh trội nhiễm sắc thể thường (Autosomal Dominant Optic Atrophy; ADOA) là một bệnh thần kinh thị giác di truyền đặc trưng bởi teo thị thần kinh tiến triển hai bên. Số OMIM là 165500. Đây là bệnh thần kinh thị giác di truyền phổ biến nhất, với tỷ lệ hiện mắc ước tính từ 1:12.000 đến 1:50.000 2).
Các bệnh thần kinh thị giác di truyền được chia thành ADOA (gen nhân, di truyền Mendel) và Bệnh thần kinh thị giác di truyền Leber (LHON) (đột biến DNA ty thể, di truyền theo dòng mẹ). Teo thị thần kinh lặn nhiễm sắc thể thường bao gồm hội chứng Wolfram. Cả ADOA và LHON đều có chung cơ chế bệnh lý thoái hóa tế bào hạch võng mạc (RGC) do rối loạn chức năng ty thể, nhưng biểu hiện lâm sàng và nguyên nhân di truyền khác nhau 6).
Tỷ lệ hiện mắc ước tính từ 1:12.000 đến 1:50.000, và đây là bệnh thần kinh thị giác di truyền phổ biến nhất 2). Tỷ lệ hiện mắc chung của các bệnh thần kinh thị giác di truyền tại Vương quốc Anh được báo cáo là khoảng 1:25.000 6).
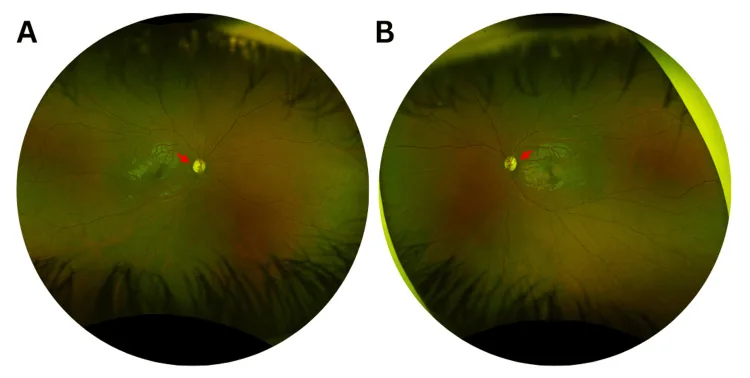
Khởi phát điển hình xảy ra ở thập kỷ đầu tiên hoặc thứ hai của cuộc đời. Do tiến triển chậm, nhiều bệnh nhân không thể xác định chính xác thời điểm khởi phát.
Có. Trong báo cáo của Tachibana và cộng sự (2025), một đột biến mới trên OPA1 đã được xác định ở một trường hợp duy trì thị lực điều chỉnh tốt nhất 0,8/0,6 ở tuổi 56 2). Tỷ lệ thâm nhập dao động từ 43–100%, và các bất thường chỉ có thể được phát hiện bằng OCT ở người mang gen không triệu chứng.
Ba dấu hiệu chính là đĩa thị giác, thị giác màu sắc và thị trường.
ADOA có kiểu hình DOA plus kèm theo các bất thường toàn thân ngoài teo thần kinh thị. 20-30% bệnh nhân có biến chứng như mất thính lực, bệnh thần kinh ngoại biên, bệnh cơ, mất điều hòa, và liệt cơ mắt ngoài tiến triển mạn tính (CPEO) 3).
Thể điển hình (DOA)
Triệu chứng: Giảm thị lực hai bên tiến triển chậm.
Dấu hiệu gai thị: Nhợt hình nêm thái dương là dấu hiệu trung tâm.
Triệu chứng toàn thân: Chỉ teo thần kinh thị.
Thị lực: Hơn 80% duy trì thị lực ≥ 0,1.
DOA plus
Tần suất: Xuất hiện ở 20-30% bệnh nhân 3).
Biến chứng: Mất thính lực, bệnh thần kinh ngoại biên, bệnh cơ, mất điều hòa, CPEO, v.v.
Thể nặng: Đột biến OPA1 hai alen gây hội chứng Behr (khởi phát sớm, suy giảm thị lực nặng, mất điều hòa, co giật) 3).
Hơn 60% trường hợp ADOA do đột biến gen OPA1 6). OPA1 nằm trên nhiễm sắc thể 3q28-q29, và hơn 500 đột biến gây bệnh đã được xác định 6). Tại Nhật Bản, đột biến c.2708_2711 delTTAG được biết đến là đột biến tần số cao.
Ở các trường hợp âm tính với OPA1, cần tìm kiếm các đột biến gen khác. Dưới đây là các gen gây bệnh chính.
| Gen | Bệnh liên quan | Ghi chú |
|---|---|---|
| OPA1 | ADOA (thể điển hình / DOA plus) | Hơn 60% tổng số 6) |
| AFG3L2 | Teo thị thần kinh type 12 (OAT12), SCA28 | Khoảng 3% bệnh thần kinh thị giác di truyền 1) |
| OPA3 | Teo thị thần kinh trội kèm đục thủy tinh thể / mất thính lực (hội chứng Costeff) | OMIM #258501 |
| WFS1 | Giống hội chứng Wolfram | OMIM #222370, #614296 |
| DNM1L | Teo thị thần kinh type 5 (OPA5) | Kiểm soát phân chia ty thể |
Brodsky và cộng sự (2023) đã xác định teo thị thần kinh type 12 do đột biến c.1064C>T (p.Thr355Met) trên gen AFG3L2 ở một người cha và con gái gốc Đông Phi (Somali)1). Trong một nghiên cứu thuần tập gồm 2186 ca, AFG3L2 nằm trong top 10 gen gây bệnh thần kinh thị giác di truyền, chiếm 14 trong số 451 ca (3%).
Có. Một số gen đã được xác định như AFG3L2, OPA3, WFS1 và DNM1L (OPA5). Đặc biệt, AFG3L2 chiếm khoảng 3% các ca bệnh thần kinh thị giác di truyền1), và khi OPA1 âm tính, việc tìm kiếm toàn diện bằng giải trình tự exome/genome là quan trọng.
Nếu phát hiện suy giảm thị lực hai bên không rõ nguyên nhân ở độ tuổi đi học, hãy nghi ngờ bệnh này. Tiền sử gia đình có triệu chứng tương tự là manh mối quan trọng, nhưng có thể không có tiền sử gia đình do tính thâm nhập không hoàn toàn.
Trong trường hợp nam 56 tuổi của Tachibana và cộng sự (2025), thị trường HFA 24-2 kích thích trắng bình thường, nhưng thị trường blue-on-yellow phát hiện giảm độ nhạy2). OCT cho thấy mỏng RNFL thái dương, CFF giảm xuống 30/31 Hz (bình thường >39 Hz).
| Bệnh | Kiểu khởi phát | Kiểu di truyền | Điểm phân biệt |
|---|---|---|---|
| LHON | Cấp đến bán cấp | Di truyền theo mẹ | Ưa thích nam trẻ, nặng |
| ADOA | Chậm và âm thầm | Di truyền trội nhiễm sắc thể thường | Thời thơ ấu, hai mắt đối xứng |
| Glôcôm | Chậm | Đa yếu tố | Tăng nhãn áp, lõm gai thị rộng |
| Bệnh thần kinh thị giác do chèn ép | Chậm đến bán cấp | Không di truyền | Tổn thương giao thoa thị giác trên MRI |
Chẩn đoán phân biệt khác: bệnh thần kinh thị giác do độc chất (ví dụ ethambutol), bệnh thần kinh thị giác do thiếu dinh dưỡng, loạn dưỡng hoàng điểm tiềm ẩn, loạn dưỡng tế bào nón, nhược thị chức năng, rối loạn thị giác tâm lý.
Không có phương pháp điều trị hiệu quả đã được thiết lập. Chăm sóc thị lực kém và tư vấn bệnh nhân là chủ yếu trong điều trị.
Để giảm stress oxy hóa, những điều sau đã được đề xuất, nhưng chưa có phương pháp nào được thiết lập như điều trị tiêu chuẩn.
Cần giải thích về di truyền trội trên nhiễm sắc thể thường và cung cấp thông tin về nguy cơ nặng do đột biến hai alen (hội chứng Behr) 3).
Suy giảm thị lực thường tiến triển chậm và nhẹ hơn so với LHON. Hơn 80% bệnh nhân duy trì thị lực chỉnh kính từ 0,1 trở lên, nhưng có trường hợp giảm xuống 0,1 hoặc thấp hơn. Vì khởi phát chậm, nhận biết có thể muộn, đôi khi được phát hiện tình cờ khi khám. Do không có liệu pháp đã được thiết lập, theo dõi định kỳ và chăm sóc thị lực kém dài hạn là quan trọng.
Hiện chưa có phương pháp điều trị hiệu quả đã được thiết lập. Chăm sóc thị lực kém là phương pháp điều trị chính. Idebenone đã được báo cáo có khả năng ổn định hoặc phục hồi thị lực 4)5), nhưng chưa được thiết lập như một phương pháp điều trị tiêu chuẩn. Đối với các phương pháp điều trị đang trong giai đoạn nghiên cứu, hãy tham khảo phần “Nghiên cứu mới nhất và Triển vọng tương lai”.
OPA1 là một GTPase liên quan đến dynamin nằm ở màng trong ty thể. Được tổng hợp trong nhân rồi vận chuyển đến ty thể, thực hiện các chức năng sau 6).
Cơ chế bệnh lý chính là bất hoạt một alen (haploinsufficiency). Hầu hết các đột biến OPA1 gây kết thúc sớm quá trình dịch mã, dẫn đến thiếu hụt lượng protein OPA1 6).
Giảm protein OPA1 → Tăng phân mảnh ty thể và tăng tái chế 3) → Rối loạn chuyển hóa ty thể và suy giảm phosphoryl hóa oxy hóa → Tăng các loại oxy phản ứng (ROS) → Chết theo chương trình của tế bào hạch võng mạc (RGC).
Chủ yếu các tế bào hạch võng mạc ở bó gai thị-hoàng điểm bị thoái hóa, biểu hiện dưới dạng teo thần kinh thị giác phía thái dương.
AFG3L2 mã hóa tiểu đơn vị của metalloprotease AAA ma trận ty thể (m-AAA) 1). Hình thành phức hợp với SPG7 (paraplegin) và thực hiện quá trình xử lý, trưởng thành và kiểm soát chất lượng protein ty thể phụ thuộc ATP. Đột biến gây thoái hóa RGC tương tự như OPA1 1).
Liệu pháp ASO
Mục tiêu: Exon cảm ứng NMD (phân hủy phụ thuộc vô nghĩa) của pre-mRNA OPA1.
Cơ chế: Ức chế sự bao gồm exon cảm ứng NMD, tăng cường dịch mã OPA1 kiểu hoang dã bằng phương pháp không phụ thuộc đột biến 6).
Tình trạng hiện tại: Đã xác nhận tăng sản xuất protein OPA1 và cải thiện năng lượng sinh học ty thể trên ba dòng tế bào có nguồn gốc từ bệnh nhân ADOA 6). Cần tiêm nội nhãn định kỳ, với nguy cơ viêm nội nhãn và viêm màng bồ đào mạn tính là thách thức.
Liệu pháp gen
Mô hình chuột: Liệu pháp gen OPA1 ngăn ngừa mất tế bào hạch võng mạc trên mô hình chuột DOA 7).
Tối ưu hóa đồng dạng: Các đồng dạng OPA1 được tối ưu hóa 1 và 7 cho thấy hiệu quả điều trị trên mô hình rối loạn chức năng ty thể 8).
Tình trạng hiện tại: Giai đoạn tiền lâm sàng.
Chỉnh sửa gen
CRISPR-Cas9: Chỉnh sửa đột biến OPA1 c.1334G>A: p.R445H trên iPSC phục hồi cân bằng nội môi ty thể 9).
Trans-splicing: Các phương pháp chỉnh sửa đột biến gây bệnh ở mức mRNA cũng đang được nghiên cứu 6).
Tế bào gốc: Tái tạo dây thần kinh thị giác bằng RGC có nguồn gốc từ iPSC đang được nghiên cứu trong các thử nghiệm tiền lâm sàng 6).
Phân tích lại lặp đi lặp lại giải trình tự exome cũng là một tiến bộ chẩn đoán quan trọng. Với sự mở rộng kiến thức di truyền, các chẩn đoán mới có thể thu được từ dữ liệu trước đây âm tính trong các xét nghiệm trước đó 1).