상염색체 우성 시신경 위축증(Autosomal Dominant Optic Atrophy; ADOA)은 양안성 진행성 시신경 위축을 특징으로 하는 유전성 시신경병증입니다. OMIM 번호는 165500입니다. 가장 빈도가 높은 유전성 시신경병증이며, 유병률은 1:12,000~1:50,000으로 추정됩니다 2).
유전성 시신경병증은 크게 핵 유전자·멘델 유전 형식의 ADOA와 미토콘드리아 DNA 변이·모계 유전의 Leber 유전성 시신경병증(LHON)으로 나뉩니다. 상염색체 열성 유전 형식의 시신경 위축에는 Wolfram 증후군이 있습니다. ADOA와 LHON은 모두 미토콘드리아 기능 장애로 인한 망막 신경절 세포(RGC) 변성을 공통 병태로 하지만, 임상상과 유전적 원인은 다릅니다 6).
Q상염색체 우성 시신경 위축증은 얼마나 자주 발생합니까?
A
유병률은 1:12,000~1:50,000으로 추정되며, 유전성 시신경병증 중에서 가장 빈도가 높습니다 2). 영국에서 유전성 시신경병증 전체의 유병률은 약 1:25,000으로 보고되었습니다 6).
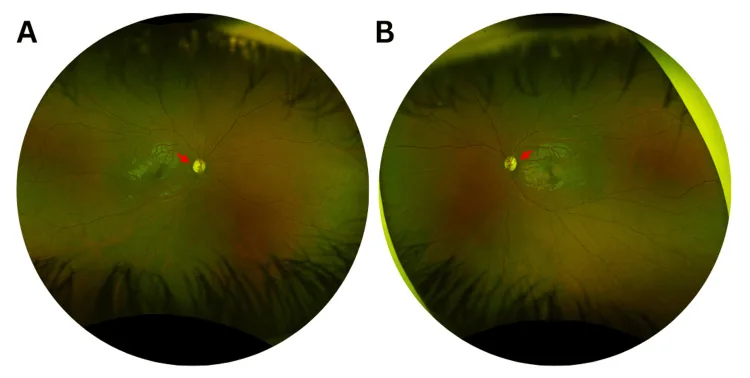
Murati Calderon RA, et al. Clinical and Genetic Findings in an Autosomal Dominant Optic Atrophy-Compatible Phenotype Harboring an OPA1 Variant: A Case Report. Cureus. 2025. Figure 1. PMCID: PMC12659938. License: CC BY.
초진 시 우안(A)과 좌안(B)의 컬러 안저 사진으로, 양안의 경미한 이측 시신경 유두 창백(빨간 화살표)과 시신경 유두 함몰 확장을 보입니다. 본문 “2. 주요 증상과 임상 소견” 항목에서 다루는 시신경 유두 창백에 해당합니다.
Brodsky 등(2023)은 동아프리카(소말리)계 부녀에서 AFG3L2 유전자 c.1064C>T(p.Thr355Met) 돌연변이로 인한 시신경 위축 12형을 확인했습니다1). 2186명의 코호트 연구에서 유전성 시신경병증의 원인 유전자 상위 10위 안에 AFG3L2가 포함되었으며, 451명 중 14명(3%)을 차지했습니다.
QOPA1 유전자 외에도 원인 유전자가 있습니까?
A
있습니다. AFG3L2, OPA3, WFS1, DNM1L(OPA5) 등 여러 유전자가 확인되었습니다. 특히 AFG3L2는 유전성 시신경병증의 약 3%를 차지하며1), OPA1이 음성인 경우 엑솜/게놈 시퀀싱을 통한 포괄적 검색이 중요합니다.
시력 저하는 일반적으로 서서히 진행되며, LHON에 비해 경미한 경과를 보입니다. 80% 이상의 환자가 교정 시력 0.1 이상을 유지하지만, 교정 시력 0.1 이하로 떨어지는 경우도 있습니다. 발병이 서서히 진행되어 자각이 늦어지고, 검진에서 우연히 발견되기도 합니다. 확립된 치료법이 없으므로 정기적인 모니터링과 저시력 관리를 통한 장기 관리가 중요합니다.
QADOA에 효과적인 치료법이 있습니까?
A
현재 확립된 효과적인 치료법은 없습니다. 저시력 관리가 치료의 주축입니다. 이데베논은 시력 안정화 및 회복 가능성을 보여주는 보고가 있지만4)5), 표준 치료로 확립되지는 않았습니다. 연구 단계의 치료 접근법에 대해서는 [“최신 연구와 향후 전망” 항목](#7-최신의 연구와 향후 전망연구 단계의 보고)을 참조하십시오.
Brodsky MC, Olson RJ, Asumda FZ, Lopour MQR, Schimmenti LA, Klee EW. Identification of AFG3L2 dominant optic atrophy following reanalysis of clinical exome sequencing. American journal of ophthalmology case reports. 2023;30:101825. doi:10.1016/j.ajoc.2023.101825. PMID:36974169; PMCID:PMC10038781.
Tachibana M, Hayashi T, Igawa Y, Mizobuchi K, Miyasaka Y, Tsuchihashi T, et al. Case of autosomal dominant optic atrophy with relatively good visual function. BMC ophthalmology. 2025;25(1):443. doi:10.1186/s12886-025-04276-5. PMID:40751186; PMCID:PMC12315315.
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes. 2022;13(6). doi:10.3390/genes13061005. PMID:35741767; PMCID:PMC9223020.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain : a journal of neurology. 2013;136(Pt 2):e231. doi:10.1093/brain/aws280. PMID:23388408.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Annals of clinical and translational neurology. 2020;7(4):590-594. doi:10.1002/acn3.51026. PMID:32243103; PMCID:PMC7187718.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Sarzi E, Seveno M, Piro-Mégy C, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep. 2018;8:2468. doi:10.1038/s41598-018-20838-8.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Frontiers in neuroscience. 2020;14:571479. doi:10.3389/fnins.2020.571479. PMID:33324145; PMCID:PMC7726421.
Sladen PE, Perdigão PRL, Salsbury G, Novoselova T, van der Spuy J, Chapple JP, et al. CRISPR-Cas9 correction of OPA1 c.1334G>A: p.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Molecular therapy. Nucleic acids. 2021;26:432-443. doi:10.1016/j.omtn.2021.08.015. PMID:34589289; PMCID:PMC8455316.