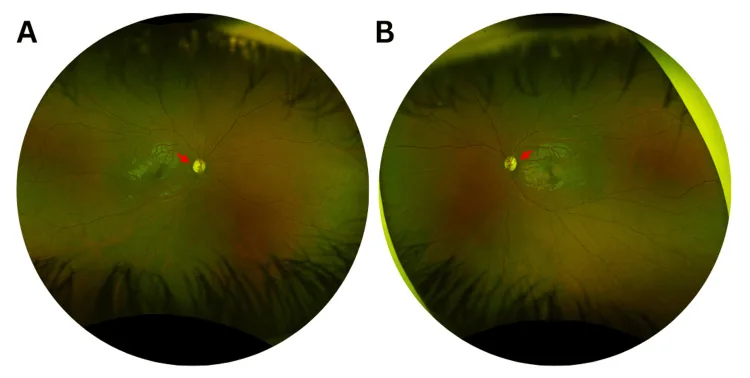
Murati Calderon RA, et al. Clinical and Genetic Findings in an Autosomal Dominant Optic Atrophy-Compatible Phenotype Harboring an OPA1 Variant: A Case Report. Cureus. 2025. Figure 1. PMCID: PMC12659938. License: CC BY.
Brodsky MC, Olson RJ, Asumda FZ, Lopour MQR, Schimmenti LA, Klee EW. Identification of AFG3L2 dominant optic atrophy following reanalysis of clinical exome sequencing. American journal of ophthalmology case reports. 2023;30:101825. doi:10.1016/j.ajoc.2023.101825. PMID:36974169; PMCID:PMC10038781.
Tachibana M, Hayashi T, Igawa Y, Mizobuchi K, Miyasaka Y, Tsuchihashi T, et al. Case of autosomal dominant optic atrophy with relatively good visual function. BMC ophthalmology. 2025;25(1):443. doi:10.1186/s12886-025-04276-5. PMID:40751186; PMCID:PMC12315315.
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes. 2022;13(6). doi:10.3390/genes13061005. PMID:35741767; PMCID:PMC9223020.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain : a journal of neurology. 2013;136(Pt 2):e231. doi:10.1093/brain/aws280. PMID:23388408.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Annals of clinical and translational neurology. 2020;7(4):590-594. doi:10.1002/acn3.51026. PMID:32243103; PMCID:PMC7187718.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Sarzi E, Seveno M, Piro-Mégy C, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep. 2018;8:2468. doi:10.1038/s41598-018-20838-8.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Frontiers in neuroscience. 2020;14:571479. doi:10.3389/fnins.2020.571479. PMID:33324145; PMCID:PMC7726421.
Sladen PE, Perdigão PRL, Salsbury G, Novoselova T, van der Spuy J, Chapple JP, et al. CRISPR-Cas9 correction of OPA1 c.1334G>A: p.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Molecular therapy. Nucleic acids. 2021;26:432-443. doi:10.1016/j.omtn.2021.08.015. PMID:34589289; PMCID:PMC8455316.