Tipo típico (DOA)
Síntomas: Pérdida visual bilateral lentamente progresiva.
Hallazgos papilares: Palidez temporal en cuña es un hallazgo central.
Síntomas sistémicos: Solo atrofia óptica.
Agudeza visual: Más del 80% mantiene 0.1 o mejor.

La Atrofia Óptica Autosómica Dominante (ADOA) es una neuropatía óptica hereditaria caracterizada por atrofia óptica progresiva bilateral. El número OMIM es 165500. Es la neuropatía óptica hereditaria más frecuente, con una prevalencia estimada de 1:12,000 a 1:50,000 2).
Las neuropatías ópticas hereditarias se dividen ampliamente en ADOA, causada por mutaciones en genes nucleares con herencia mendeliana, y la neuropatía óptica hereditaria de Leber (LHON), causada por mutaciones en el ADN mitocondrial con herencia materna. La atrofia óptica autosómica recesiva incluye el síndrome de Wolfram. Tanto la ADOA como la LHON comparten la patología común de degeneración de las células ganglionares de la retina (RGC) debido a disfunción mitocondrial, pero sus características clínicas y causas genéticas difieren 6).
La prevalencia se estima en 1:12,000 a 1:50,000, siendo la neuropatía óptica hereditaria más común 2). En el Reino Unido, la prevalencia general de neuropatías ópticas hereditarias se reporta en aproximadamente 1:25,000 6).
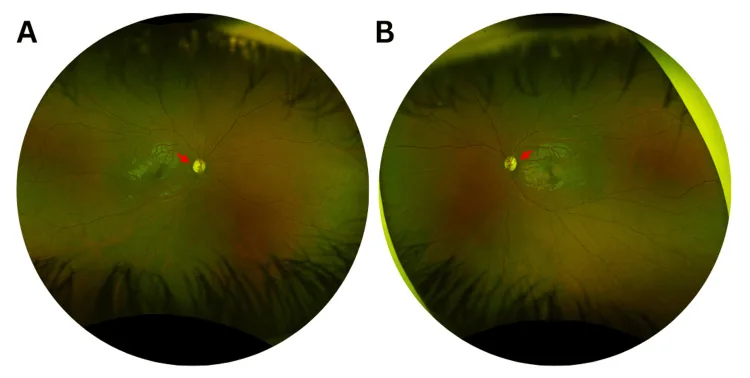
La aparición típica ocurre en la primera o segunda década de la vida. Debido a que la progresión es lenta, muchos pacientes no pueden precisar el momento exacto de inicio.
Sí. En un informe de Tachibana et al. (2025), se identificó una nueva mutación en OPA1 en un paciente que mantuvo una agudeza visual corregida de 0.8/0.6 a los 56 años 2). La penetrancia varía del 43% al 100%, y las anomalías pueden detectarse solo mediante OCT en portadores asintomáticos.
Los tres hallazgos principales involucran el disco óptico, la visión del color y el campo visual.
La ADOA puede presentar un fenotipo DOA plus con anomalías sistémicas además de la atrofia óptica. El 20-30% de los pacientes presentan complicaciones como pérdida auditiva, neuropatía periférica, miopatía, ataxia y oftalmoplejía externa progresiva crónica (CPEO) 3).
Tipo típico (DOA)
Síntomas: Pérdida visual bilateral lentamente progresiva.
Hallazgos papilares: Palidez temporal en cuña es un hallazgo central.
Síntomas sistémicos: Solo atrofia óptica.
Agudeza visual: Más del 80% mantiene 0.1 o mejor.
DOA plus
Frecuencia: Ocurre en el 20-30% de los pacientes 3).
Complicaciones: Pérdida auditiva, neuropatía periférica, miopatía, ataxia, CPEO, etc.
Tipo grave: Las mutaciones bialélicas de OPA1 causan el síndrome de Behr (inicio temprano, deterioro visual severo, ataxia, espasticidad) 3).
Más del 60% de los casos de ADOA son causados por mutaciones en el gen OPA1 6). OPA1 se localiza en el cromosoma 3q28-q29 y se han identificado más de 500 mutaciones patogénicas 6). En Japón, c.2708_2711 delTTAG se conoce como una mutación frecuente.
En casos negativos para OPA1, se deben buscar otras mutaciones genéticas. Los principales genes causantes se muestran a continuación.
| Gen | Enfermedad asociada | Notas |
|---|---|---|
| OPA1 | ADOA (tipo típico, DOA plus) | Más del 60% del total6) |
| AFG3L2 | Atrofia óptica tipo 12 (OAT12), SCA28 | Aproximadamente el 3% de las neuropatías ópticas hereditarias1) |
| OPA3 | Atrofia óptica dominante con cataratas y pérdida auditiva (síndrome de Costeff) | OMIM #258501 |
| WFS1 | Síndrome de Wolfram-like | OMIM #222370, #614296 |
| DNM1L | Atrofia óptica tipo 5 (OPA5) | Regulación de la fisión mitocondrial |
Brodsky et al. (2023) identificaron atrofia óptica tipo 12 debida a la mutación c.1064C>T (p.Thr355Met) en el gen AFG3L2 en un padre y una hija de ascendencia africana oriental (somalí) 1). En un estudio de cohorte de 2186 casos, AFG3L2 se encontraba entre los 10 principales genes causantes de neuropatía óptica hereditaria, representando 14 de 451 casos (3%).
Sí. Se han identificado varios genes, incluyendo AFG3L2, OPA3, WFS1 y DNM1L (OPA5). En particular, AFG3L2 representa aproximadamente el 3% de las neuropatías ópticas hereditarias 1), y cuando OPA1 es negativo, es importante realizar una búsqueda exhaustiva mediante secuenciación de exoma/genoma.
Sospeche esta enfermedad cuando se descubra un trastorno bilateral del desarrollo visual de causa desconocida durante la edad escolar. Los antecedentes familiares de síntomas similares son una pista importante, pero debido a la penetrancia incompleta, puede no haber antecedentes familiares.
En un caso de un varón de 56 años reportado por Tachibana et al. (2025), la perimetría HFA 24-2 con estímulo blanco fue normal, pero la perimetría azul-amarillo detectó pérdida de sensibilidad2). La OCT mostró adelgazamiento de la RNFL temporal, y la CFF se redujo a 30/31 Hz (normal >39 Hz).
| Enfermedad | Forma de inicio | Herencia | Puntos clave de diferenciación |
|---|---|---|---|
| LHON | Agudo a subagudo | Herencia materna | Predominio en varones jóvenes, grave |
| ADOA | Lento, insidioso | Autosómico dominante | Infancia, bilateral simétrico |
| Glaucoma | Lento | Multifactorial | Elevación de la presión intraocular, excavación papilar aumentada |
| Neuropatía óptica compresiva | Lento a subagudo | No hereditario | RM muestra lesión quiasmática |
Otros diagnósticos diferenciales: neuropatía óptica tóxica (p. ej., etambutol), neuropatía óptica por deficiencia nutricional, distrofia macular oculta, distrofia de conos, ambliopía funcional, trastorno visual psicógeno.
No existe un tratamiento eficaz establecido. El cuidado de la baja visión y el asesoramiento al paciente son los pilares del tratamiento.
Se han propuesto los siguientes para reducir el estrés oxidativo, pero ninguno está establecido como tratamiento estándar.
Es importante explicar la herencia autosómica dominante y proporcionar información sobre el riesgo de enfermedad grave (síndrome de Behr) debido a mutaciones bialélicas 3).
La pérdida de visión generalmente progresa lentamente y sigue un curso más leve en comparación con la LHON. Más del 80% de los pacientes mantienen una agudeza visual corregida de 0.1 o mejor, pero algunos casos disminuyen a 0.1 o peor. Debido a que el inicio es gradual, los pacientes pueden no notarlo y, a veces, se descubre incidentalmente durante los chequeos. Dado que no existe un tratamiento establecido, es importante el manejo a largo plazo con monitoreo regular y cuidado de baja visión.
Actualmente no existe un tratamiento eficaz establecido. El cuidado de la baja visión es el pilar del tratamiento. La idebenona ha mostrado potencial para estabilizar o mejorar la visión en algunos informes4)5), pero no está establecida como tratamiento estándar. Para los enfoques de tratamiento en fase de investigación, consulte la sección “Investigación más reciente y perspectivas futuras”.
OPA1 es una GTPasa relacionada con la dinamina ubicada en la membrana interna mitocondrial. Se sintetiza en el núcleo y se transporta a las mitocondrias, donde realiza las siguientes funciones6).
El principal mecanismo patológico es la haploinsuficiencia. La mayoría de las mutaciones de OPA1 causan terminación prematura de la traducción, lo que lleva a una cantidad insuficiente de proteína OPA16).
Disminución de la proteína OPA1 → aumento de la fragmentación mitocondrial y reciclaje mejorado3) → disfunción metabólica mitocondrial y alteración de la fosforilación oxidativa → elevación de especies reactivas de oxígeno (ROS) → apoptosis de las CGR.
Las CGR del haz papilomacular se ven afectadas principalmente, manifestándose como atrofia óptica temporal.
AFG3L2 codifica una subunidad de la metaloproteasa AAA de la matriz mitocondrial (m-AAA)1). Forma un complejo con SPG7 (paraplegina) y realiza el procesamiento, maduración y control de calidad de las proteínas mitocondriales de manera dependiente de ATP. Las mutaciones causan degeneración de las CGR similar a OPA11).
Terapia ASO
Diana: Exón inductor de NMD (degradación mediada por codones sin sentido) del pre-ARNm de OPA1.
Mecanismo: Inhibe la inclusión del exón que induce NMD, potenciando la traducción de OPA1 de tipo salvaje mediante un enfoque independiente de mutación 6).
Estado actual: Se confirmó el aumento de la producción de proteína OPA1 y la mejora de la bioenergética mitocondrial en tres líneas celulares derivadas de pacientes con ADOA 6). Requiere administración repetida regular mediante inyección intravítrea, con riesgos de endoftalmitis y uveítis crónica como desafíos.
Terapia génica
Modelo murino: La terapia génica con OPA1 previno la pérdida de RGC en un modelo murino de DOA 7).
Optimización de isoformas: Las isoformas optimizadas 1 y 7 de OPA1 muestran efectos terapéuticos en modelos de disfunción mitocondrial 8).
Estado actual: Etapa preclínica.
Edición génica
CRISPR-Cas9: La corrección de la mutación OPA1 c.1334G>A: p.R445H en iPSC restauró la homeostasis mitocondrial 9).
Trans-empalme: También se investiga un enfoque para corregir mutaciones patogénicas a nivel de ARNm 6).
Células madre: Se están investigando en estudios preclínicos la regeneración del nervio óptico mediante RGC derivadas de iPSC6).
El reanálisis iterativo de la secuenciación del exoma también es un avance diagnóstico importante. Con la expansión del conocimiento genético, se pueden obtener nuevos diagnósticos a partir de datos que antes eran negativos1).