Klasik tip (DOA)
Belirtiler: Bilateral yavaş ilerleyici görme azalması.
Papil bulguları: Temporal kama şeklinde solukluk temel bulgudur.
Sistemik belirtiler: Yalnızca optik atrofi.
Görme keskinliği: %80’den fazla hastada 0.1 veya daha iyidir.

Otozomal dominant optik atrofi (ADOA), iki taraflı ilerleyici optik sinir atrofisi ile karakterize kalıtsal bir optik nöropatidir. OMIM numarası 165500’dür. En sık görülen kalıtsal optik nöropatidir ve prevalansı 1:12.000 ila 1:50.000 arasında tahmin edilmektedir2).
Kalıtsal optik nöropatiler temel olarak ikiye ayrılır: nükleer genler ve Mendel kalıtımı ile ADOA ve mitokondriyal DNA mutasyonları ve maternal kalıtım ile Leber kalıtsal optik nöropatisi (LHON). Otozomal resesif optik atrofi Wolfram sendromunu içerir. Hem ADOA hem de LHON, ortak patoloji olarak mitokondriyal disfonksiyona bağlı retinal ganglion hücre (RGC) dejenerasyonuna sahiptir, ancak klinik tablo ve genetik nedenler farklıdır6).
Prevalansı 1:12.000 ila 1:50.000 arasında tahmin edilmektedir ve kalıtsal optik nöropatiler arasında en sık görülenidir2). Birleşik Krallık’ta tüm kalıtsal optik nöropatilerin prevalansı yaklaşık 1:25.000 olarak bildirilmiştir6).
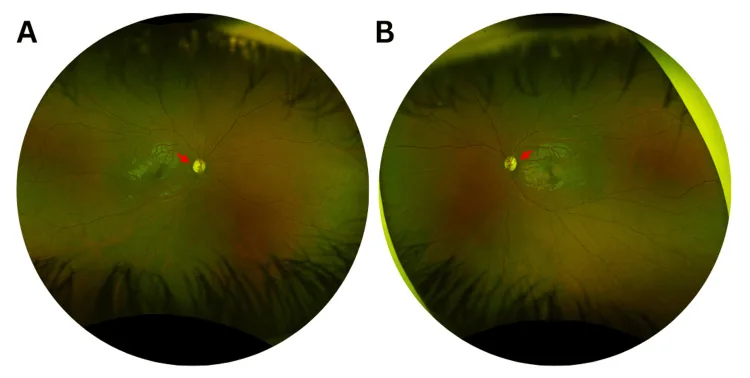
Tipik başlangıç yaşamın 1. ve 2. dekatlarındadır. Yavaş ilerlediği için birçok hasta kesin başlangıç zamanını belirleyemez.
Evet. Tachibana ve ark. (2025) raporunda, 56 yaşında en iyi düzeltilmiş görme keskinliği 0.8/0.6 olan bir olguda OPA1’de yeni bir mutasyon tanımlandı 2). Penetrans %43-100 arasında değişir ve asemptomatik taşıyıcılarda bile sadece OCT ile anormallik saptanabilir.
Üç ana bulgu: Optik sinir başı, Renk görme ve Görme alanı.
ADOA’da optik atrofi dışında sistemik anormalliklerin eşlik ettiği DOA plus fenotipi bulunur. Hastaların %20-30’unda işitme kaybı, periferik nöropati, miyopati, ataksi, kronik progresif eksternal oftalmopleji (CPEO) gibi ek bulgular görülür3).
Klasik tip (DOA)
Belirtiler: Bilateral yavaş ilerleyici görme azalması.
Papil bulguları: Temporal kama şeklinde solukluk temel bulgudur.
Sistemik belirtiler: Yalnızca optik atrofi.
Görme keskinliği: %80’den fazla hastada 0.1 veya daha iyidir.
DOA plus
Sıklık: Hastaların %20-30’unda görülür3).
Komplikasyonlar: İşitme kaybı, periferik nöropati, miyopati, ataksi, CPEO vb.
Şiddetli tip: Biallelik OPA1 mutasyonları Behr sendromuna (erken başlangıç, ciddi görme bozukluğu, ataksi, konvülsiyon) neden olur3).
ADOA olgularının %60’ından fazlası OPA1 gen mutasyonundan kaynaklanır6). OPA1, kromozom 3q28-q29’da yer alır ve 500’den fazla patojenik mutasyon tanımlanmıştır6). Japonya’da c.2708_2711 delTTAG sık görülen bir mutasyondur.
OPA1 negatif vakalarda diğer gen mutasyonları araştırılmalıdır. Başlıca neden genler aşağıda verilmiştir.
| Gen | İlişkili hastalık | Notlar |
|---|---|---|
| OPA1 | ADOA (klasik tip ve DOA plus) | Toplamın %60’ından fazlası6) |
| AFG3L2 | Optik atrofi tip 12 (OAT12), SCA28 | Kalıtsal optik nöropatilerin yaklaşık %3’ü1) |
| OPA3 | Dominant optik atrofi + katarakt ve işitme kaybı (Costeff sendromu) | OMIM #258501 |
| WFS1 | Wolfram sendromu benzeri | OMIM #222370, #614296 |
| DNM1L | Optik atrofi tip 5 (OPA5) | Mitokondriyal bölünme kontrolü |
Brodsky ve ark. (2023), Doğu Afrika (Somali) kökenli bir baba ve kızında AFG3L2 genindeki c.1064C>T (p.Thr355Met) mutasyonuna bağlı optik atrofi tip 12’yi tanımladı1). 2186 vakalık bir kohort çalışmasında, kalıtsal optik nöropatinin en sık 10 geni arasında AFG3L2 yer aldı ve 451 vakanın 14’ünde (%3) bulundu.
Evet. AFG3L2, OPA3, WFS1, DNM1L (OPA5) gibi birden fazla gen tanımlanmıştır. Özellikle AFG3L2, kalıtsal optik nöropatilerin yaklaşık %3’ünü oluşturur1) ve OPA1 negatif olduğunda ekzom/genom dizileme ile kapsamlı arama önemlidir.
Okul çağında keşfedilen, nedeni bilinmeyen iki taraflı görme gelişim bozukluğu görüldüğünde bu hastalıktan şüphelenilir. Benzer semptomların aile öyküsü önemli bir ipucudur, ancak eksik penetrans nedeniyle aile öyküsü olmayabilir.
Tachibana ve ark. (2025) tarafından bildirilen 56 yaşındaki erkek olguda, HFA 24-2 beyaz uyaranlı perimetri normaldi ancak mavi-üzerine-sarı perimetride hassasiyet kaybı saptandı2). OCT temporal RNFL incelmesi gösterdi ve CFF 30/31 Hz (normal >39 Hz) olarak düşük bulundu.
| Hastalık | Başlangıç şekli | Kalıtım şekli | Ayırıcı noktalar |
|---|---|---|---|
| LHON | Akut-subakut | Maternal kalıtım | Genç erkeklerde sık, şiddetli |
| ADOA | Yavaş ve sinsi | Otozomal dominant | Çocukluk çağı, iki taraflı simetrik |
| Glokom | Yavaş | Multifaktöriyel | Göz içi basıncı artışı, optik disk çukurlaşması |
| Basıya bağlı optik nöropati | Yavaştan subakuta | Kalıtsal olmayan | MRG’de kiazmal lezyon |
Diğer ayırıcı tanılar: Toksik optik nöropati (etambutol vb.), beslenme yetersizliğine bağlı optik nöropati, okült maküler distrofi, koni distrofisi, fonksiyonel ambliyopi, psikojenik görme bozukluğu.
Kanıtlanmış etkili bir tedavi yoktur. Az görme rehabilitasyonu ve hasta danışmanlığı tedavinin temelini oluşturur.
Oksidatif stresi azaltmak amacıyla aşağıdakiler önerilmiştir, ancak hiçbiri standart tedavi olarak kabul edilmemiştir.
Otozomal dominant kalıtımın açıklanması ve biallelik mutasyonlara bağlı şiddetli hastalık (Behr sendromu) riski hakkında bilgi verilmesi önemlidir 3).
Görme kaybı genellikle yavaş ilerler ve LHON’a kıyasla daha hafif bir seyir izler. Hastaların %80’inden fazlası düzeltilmiş görme keskinliğini 0.1 veya üzerinde korur, ancak 0.1’in altına düşen vakalar da vardır. Başlangıcın yavaş olması nedeniyle fark edilmeyebilir ve muayenelerde tesadüfen saptanabilir. Kesin bir tedavi olmadığından, düzenli izlem ve az görme bakımı ile uzun süreli yönetim önemlidir.
Şu anda kanıtlanmış etkili bir tedavi yoktur. Tedavinin temelini az görme rehabilitasyonu oluşturur. İdebenonun görme keskinliğini stabilize etme veya iyileştirme olasılığına dair raporlar bulunmakla birlikte4)5), standart tedavi olarak kabul edilmemiştir. Araştırma aşamasındaki tedavi yaklaşımları için “Güncel Araştırmalar ve Gelecek Perspektifleri” bölümüne bakınız.
OPA1, mitokondri iç zarında bulunan bir dinamin benzeri GTPaz’dır. Çekirdekte sentezlendikten sonra mitokondriye taşınır ve aşağıdaki işlevleri yerine getirir6):
Ana patolojik mekanizma haployetersizliktir. OPA1 mutasyonlarının çoğu translasyonun erken sonlanmasına neden olur ve OPA1 proteini miktarında azalmaya yol açar6).
OPA1 proteini azalması → Mitokondriyal parçalanmada artış ve geri dönüşümde hızlanma3) → Mitokondriyal metabolizma bozukluğu ve oksidatif fosforilasyon hasarı → Reaktif oksijen türlerinde (ROS) artış → RGC’lerin apoptozu.
Başlıca papillomaküler demetteki RGC’ler dejenere olur ve temporal optik atrofi olarak ortaya çıkar.
AFG3L2, mitokondriyal matriks AAA metaloproteazının (m-AAA) bir alt birimini kodlar1). SPG7 (paraplegin) ile kompleks oluşturarak mitokondriyal proteinlerin işlenmesini, olgunlaşmasını ve kalite kontrolünü ATP’ye bağımlı olarak gerçekleştirir. Mutasyonları, OPA1’e benzer şekilde RGC dejenerasyonuna neden olur1).
ASO Tedavisi
Hedef: OPA1 pre-mRNA’sının NMD (anlamsız aracılı bozunma) indükleyici ekzonu.
Mekanizma: NMD’yi indükleyen ekzonun eklenmesini engelleyerek, mutasyondan bağımsız bir yaklaşımla vahşi tip OPA1 translasyonunu artırma 6).
Mevcut Durum: Üç ADOA hasta kaynaklı hücre hattında OPA1 protein üretiminde artış ve mitokondriyal biyoenerjetikte iyileşme doğrulanmıştır 6). Vitre içi enjeksiyonla düzenli tekrarlanan dozlar gereklidir; endoftalmi ve kronik üveit riski zorluk oluşturmaktadır.
Gen Tedavisi
Fare Modeli: OPA1 gen tedavisi, DOA fare modelinde RGC kaybını önler 7).
İzoform Optimizasyonu: Optimize edilmiş OPA1 izoformları 1 ve 7, mitokondriyal disfonksiyon modellerinde terapötik etki gösterir 8).
Mevcut Durum: Klinik öncesi aşama.
Gen Düzenleme
CRISPR-Cas9: OPA1 c.1334G>A: p.R445H mutasyonunun iPSC’de düzeltilmesi mitokondriyal homeostazı geri kazandırır 9).
Trans-splicing: mRNA düzeyinde patojenik mutasyonları düzeltmeye yönelik yaklaşımlar da araştırılmaktadır 6).
Kök hücreler: iPSC kaynaklı RGC’ler ile optik sinir rejenerasyonu preklinik çalışmalarda araştırılmaktadır6).
Eksom dizilemenin tekrarlayan yeniden analizi de tanısal açıdan önemli bir ilerlemedir. Genetik bilginin genişlemesiyle birlikte, önceki testlerde negatif olan verilerden yeni tanılar elde edilebilir1).