Typical type (DOA)
Symptoms: Bilateral slowly progressive visual loss.
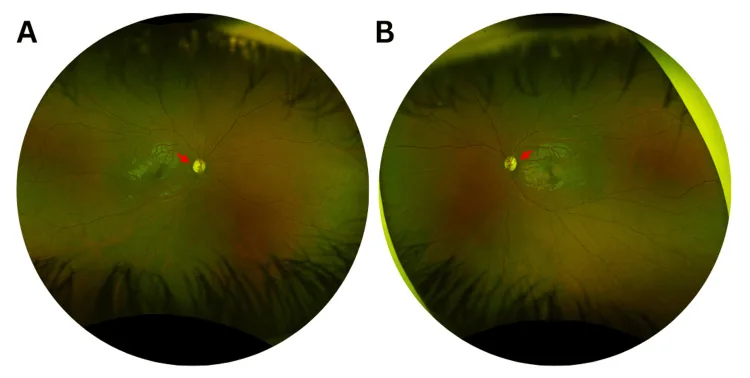
Optic disc findings: Temporal wedge pallor is a key finding.
Systemic symptoms: Optic atrophy only.
Visual acuity: Over 80% maintain 0.1 or better.

Autosomal Dominant Optic Atrophy (ADOA) is a hereditary optic neuropathy characterized by bilateral progressive optic atrophy. OMIM number is 165500. It is the most common hereditary optic neuropathy, with an estimated prevalence of 1:12,000 to 1:50,000 2).
Hereditary optic neuropathies are broadly divided into ADOA, caused by nuclear gene mutations with Mendelian inheritance, and Leber hereditary optic neuropathy (LHON), caused by mitochondrial DNA mutations with maternal inheritance. Autosomal recessive optic atrophy includes Wolfram syndrome. Both ADOA and LHON share the common pathology of retinal ganglion cell (RGC) degeneration due to mitochondrial dysfunction, but their clinical features and genetic causes differ 6).
The prevalence is estimated at 1:12,000 to 1:50,000, making it the most common hereditary optic neuropathy 2). In the UK, the overall prevalence of hereditary optic neuropathies is reported to be about 1:25,000 6).
Typical onset occurs in the first to second decades of life. Because progression is slow, many patients cannot pinpoint the exact time of onset.
Yes. In a report by Tachibana et al. (2025), a novel OPA1 mutation was identified in a patient who maintained best-corrected visual acuity of 0.8/0.6 at age 56 2). Penetrance ranges from 43% to 100%, and abnormalities may be detected only by OCT in asymptomatic carriers.
The three main findings involve the optic disc, color vision, and visual field.
ADOA can present as DOA plus phenotype with systemic abnormalities beyond optic atrophy. 20–30% of patients have complications such as hearing loss, peripheral neuropathy, myopathy, ataxia, and chronic progressive external ophthalmoplegia (CPEO) 3).
Typical type (DOA)
Symptoms: Bilateral slowly progressive visual loss.
Optic disc findings: Temporal wedge pallor is a key finding.
Systemic symptoms: Optic atrophy only.
Visual acuity: Over 80% maintain 0.1 or better.
DOA plus
Frequency: Occurs in 20–30% of patients 3).
Complications: Hearing loss, peripheral neuropathy, myopathy, ataxia, CPEO, etc.
Severe type: Biallelic OPA1 mutations cause Behr syndrome (early onset, severe visual impairment, ataxia, spasticity) 3).
Over 60% of ADOA cases are caused by OPA1 gene mutations 6). OPA1 is located on chromosome 3q28-q29, and more than 500 pathogenic mutations have been identified 6). In Japan, c.2708_2711 delTTAG is known as a frequent mutation.
In OPA1-negative cases, other genetic mutations should be investigated. The main causative genes are listed below.
| Gene | Associated disease | Notes |
|---|---|---|
| OPA1 | ADOA (typical type, DOA plus) | Over 60% of all cases6) |
| AFG3L2 | Optic atrophy type 12 (OAT12), SCA28 | Approximately 3% of hereditary optic neuropathies1) |
| OPA3 | Dominant optic atrophy with cataract and hearing loss (Costeff syndrome) | OMIM #258501 |
| WFS1 | Wolfram syndrome-like | OMIM #222370, #614296 |
| DNM1L | Optic atrophy type 5 (OPA5) | Mitochondrial fission regulation |
Brodsky et al. (2023) identified optic atrophy type 12 due to the AFG3L2 gene c.1064C>T (p.Thr355Met) mutation in a father and daughter of East African (Somali) descent 1). In a cohort study of 2186 cases, AFG3L2 was among the top 10 causative genes for hereditary optic neuropathy, accounting for 14 of 451 cases (3%).
Yes. Several genes have been identified, including AFG3L2, OPA3, WFS1, and DNM1L (OPA5). In particular, AFG3L2 accounts for about 3% of hereditary optic neuropathies 1), and when OPA1 is negative, comprehensive search by exome/genome sequencing is important.
Suspect this disease when bilateral unexplained visual developmental impairment is discovered during school age. A family history of similar symptoms is an important clue, but due to incomplete penetrance, there may be no family history.
In a 56-year-old male case reported by Tachibana et al. (2025), HFA 24-2 white-on-white perimetry was normal, but blue-on-yellow perimetry detected sensitivity loss2). OCT showed temporal RNFL thinning, and CFF was reduced to 30/31 Hz (normal >39 Hz).
| Disease | Onset Pattern | Inheritance | Key Differentiating Features |
|---|---|---|---|
| LHON | Acute to subacute | Maternal inheritance | Predominantly affects young males, severe |
| ADOA | Slow, insidious | Autosomal dominant | Childhood, bilateral symmetric |
| Glaucoma | Slow | Multifactorial | Elevated intraocular pressure, enlarged optic cup |
| Compressive optic neuropathy | Slow to subacute | Non-hereditary | MRI shows chiasmal lesion |
Other differential diagnoses: toxic optic neuropathy (e.g., ethambutol), nutritional deficiency optic neuropathy, occult macular dystrophy, cone dystrophy, functional amblyopia, psychogenic visual disturbance.
There is no established effective treatment. Low vision care and patient counseling are the mainstays of treatment.
The following have been proposed to reduce oxidative stress, but none are established as standard treatment.
It is important to explain autosomal dominant inheritance and provide information on the risk of severe disease (Behr syndrome) due to biallelic mutations 3).
Vision loss generally progresses slowly and follows a milder course compared to LHON. Over 80% of patients maintain corrected visual acuity of 0.1 or better, but some cases decline to 0.1 or worse. Because onset is gradual, patients may not notice it, and it is sometimes discovered incidentally during checkups. Since there is no established treatment, long-term management with regular monitoring and low vision care is important.
Currently, there is no established effective treatment. Low vision care is the mainstay of treatment. Idebenone has been reported to show potential for stabilizing or improving vision4)5), but it is not established as a standard treatment. For investigational treatment approaches, see “Latest Research and Future Prospects” section.
OPA1 is a dynamin-related GTPase located in the mitochondrial inner membrane. It is synthesized in the nucleus and transported to mitochondria, where it performs the following functions6).
The main pathological mechanism is haploinsufficiency. Most OPA1 mutations cause premature termination of translation, leading to insufficient OPA1 protein levels6).
Decreased OPA1 protein → increased mitochondrial fragmentation and enhanced recycling3) → mitochondrial metabolic dysfunction and impaired oxidative phosphorylation → elevated reactive oxygen species (ROS) → apoptosis of RGCs.
RGCs in the papillomacular bundle are primarily affected, manifesting as temporal optic atrophy.
AFG3L2 encodes a subunit of the mitochondrial matrix AAA metalloprotease (m-AAA)1). It forms a complex with SPG7 (paraplegin) and performs ATP-dependent processing, maturation, and quality control of mitochondrial proteins. Mutations cause RGC degeneration similar to OPA11).
ASO Therapy
Target: NMD (nonsense-mediated decay)-inducing exon of OPA1 pre-mRNA.
Mechanism: Inhibits the inclusion of the exon that induces NMD, enhancing wild-type OPA1 translation through a mutation-independent approach 6).
Current Status: Confirmed increased OPA1 protein production and improved mitochondrial bioenergetics in three ADOA patient-derived cell lines 6). Requires regular repeated administration via intravitreal injection, with risks of endophthalmitis and chronic uveitis as challenges.
Gene Therapy
Mouse Model: OPA1 gene therapy prevented RGC loss in a DOA mouse model 7).
Isoform Optimization: Optimized OPA1 isoforms 1 and 7 show therapeutic effects in mitochondrial dysfunction models 8).
Current Status: Preclinical stage.
Gene Editing
CRISPR-Cas9: Correction of the OPA1 c.1334G>A: p.R445H mutation in iPSCs restored mitochondrial homeostasis 9).
Trans-splicing: An approach to correct pathogenic mutations at the mRNA level is also under investigation 6).
Stem cells: Preclinical studies are investigating optic nerve regeneration using iPSC-derived RGCs6).
Iterative reanalysis of exome sequencing is also an important diagnostic advance. With the expansion of genetic knowledge, new diagnoses may be obtained from data that were previously negative1).